Composición: Cada comprimido recubierto contiene: Dolutegravir 50 mg. Excipientes: Manitol, Povidona, Glicolato Sódico de Almidón de Papa, Celulosa Microcristalina, Dióxido de Silicio Coloidal, Estearil Fumarato de Sodio, Alcohol Polivinílico parcialmente hidrolizado, Macrogol, Talco, Dióxido de titanio, Óxido de Hierro Rojo, Óxido de Hierro Amarillo; c.s.
Indicaciones: Santigrase está indicado en combinación con otros agentes antirretrovirales para el tratamiento de la infección por el virus de la inmunodeficiencia humana tipo 1 (HIV-1).
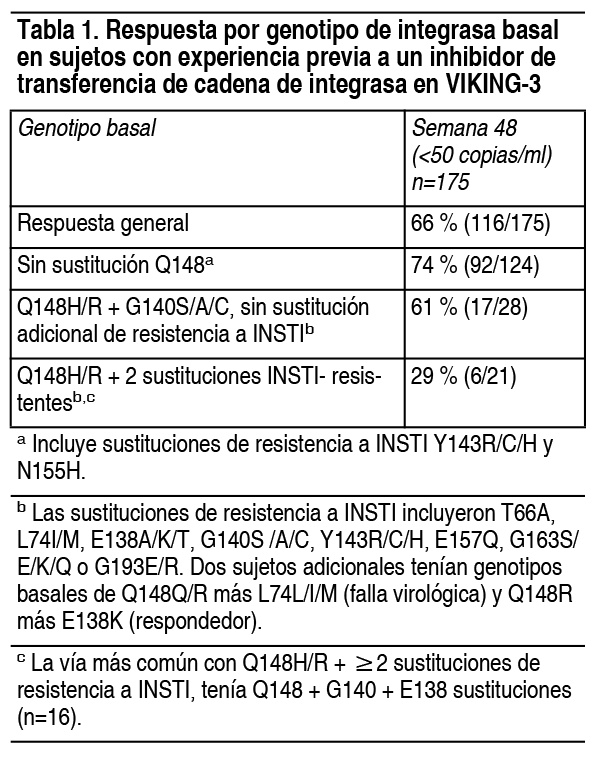
Propiedades:Farmacodinamia: Efectos sobre el electrocardiograma: En un ensayo cruzado aleatorizado, controlado con placebo, 42 sujetos sanos recibieron por vía oral una dosis única de placebo, suspensión de dolutegravir de 250 mg (exposición de, aproximadamente, 3 veces la dosis de 50 mg, 1 vez al día, en estado estable) y moxifloxacino 400 mg (control activo) en secuencia aleatoria. Después del ajuste basal y del placebo, el cambio medio máximo en el QTc basado en el método de corrección de Fridericia (QTcF) para dolutegravir fue de 2.4 ms (CI unilateral 95 %: 4.9 ms). Dolutegravir no prolongó el intervalo QTc más de 24 horas después de la dosis. Efectos sobre la función renal: El efecto de dolutegravir sobre la función renal fue evaluado en un ensayo abierto, aleatorizado, de 3 grupos, paralelo, controlado con placebo, en sujetos sanos (n=37) que recibieron 50 mg de dolutegravir, 1 vez al día (n=12), dolutegravir 50 mg, 2 veces al día (n=13) o placebo, 1 vez al día (n=12), durante 14 días. Se observó una disminución en el aclaramiento de creatinina, a través de la determinación en orina de 24 horas, con ambas dosis de dolutegravir después de 14 días de tratamiento, en sujetos que recibieron 50 mg, 1 vez al día (disminución del 9 %) y 50 mg, 2 veces al día (disminución del 13 %). Ninguna de las dosis de dolutegravir tuvo un efecto significativo sobre la tasa de filtración glomerular real (determinada por el aclaramiento del fármaco explorador iohexol) o el flujo plasmático renal efectivo (determinado por el aclaramiento del fármaco explorador para-aminohipurato), en comparación con el placebo. Microbiología: Mecanismo de acción: Dolutegravir es un agente antiviral VIH-1. Dolutegravir inhibe la integrasa del VIH al unirse al sitio activo de la enzima y bloquear el paso de transferencia de cadena de la integración del ácido desoxirribonucleico (ADN) retroviral, que es esencial para el ciclo de replicación del virus. Los ensayos bioquímicos de transferencia de cadena utilizando integrasa purificada de VIH-1 y ADN de sustrato preprocesado dieron como resultado valores de IC50 de 2.7 nM y 12.6 nM. Actividad antiviral en cultivo celular: Dolutegravir mostró actividad antiviral contra cepas de laboratorio de VIH-1 de tipo salvaje con valores medios de EC50 de 0.5 nM (0.21 ng/ml) a 2.1 nM (0.85 ng/ml) en células mononucleares de sangre periférica (PBMC) y células MT-4. Dolutegravir exhibió actividad antiviral contra 13 aislados del lado B clínicamente diversos, con un valor medio de EC50 de 0.52 nM en un ensayo de susceptibilidad a la integrasa viral, usando la región de codificación de integrasa de aislados clínicos. Dolutegravir demostró actividad antiviral en cultivo celular contra un panel de aislados clínicos de VIH-1 (3 en cada grupo de M, lados A, B, C, D, E, F y G, y 3 en el grupo O) con valores de EC50 que van desde 0.02 nM a 2.14 nM para VIH-1. Los valores C50 de dolutegravir frente a 3 aislados clínicos de VIH-2 en ensayos PBMC variaron de 0.09 nM a 0.61 nM. Actividad antiviral en combinación con otros agentes antivirales: La actividad antiviral de dolutegravir no fue antagónica cuando se combinó con el INSTI, raltegravir; con inhibidores de la transcriptasa inversa no nucleósidos (NNRTI), efavirenz o nevirapina; con inhibidores de la transcriptasa inversa nucleósidos (INTI), abacavir o estavudina; con inhibidores de la proteasa (IP), amprenavir o lopinavir; con el antagonista del correceptor CCR5, maraviroc; o el inhibidor de fusión, enfuvirtida. La actividad antiviral de dolutegravir no fue antagónica cuando se combinó con el inhibidor de la transcriptasa inversa del VHB, adefovir, o cuando se inhibió con el antiviral, ribavirina. Resistencia: Cultivo celular: Se seleccionaron virus resistentes a dolutegravir en cultivo celular a partir de diferentes cepas y dados de VIH-1 de tipo salvaje. Las sustituciones de aminoácidos E92Q, G118R, S153F o Y, G193E o R263K surgieron en diferentes pasajes y confirieron una susceptibilidad reducida a dolutegravir de hasta 4 veces y pasaje de virus mutantes que contienen las sustituciones Q148R o Q148H, seleccionadas para sustituciones adicionales en integrasa, que confieren una susceptibilidad disminuida a dolutegravir (aumento de cambio de pliegue de 13 a 46). Las sustituciones de integrasa adicionales incluyeron T97A, E138K, Gl40S y Ml54I y pasaje de virus mutantes que contienen G140S y Q148H seleccionados para L74M, E92Q y N155H. Sujetos sin tratamiento previo: Ningún sujeto en los grupos de tratamiento de 50 mg de dolutegravir, 1 vez al día, en los ensayos sin tratamiento SPRING-2 (96 semanas) y SINGLE (144 semanas), tuvo una disminución detectable en la susceptibilidad a dolutegravir o NRTI de fondo en la resistencia, subconjunto de análisis (n=12 con ARN del VIH-1 mayor de 400 copias por ml al fracaso o última visita y con datos de resistencia). Dos sujetos con insuficiencia virológica en SINGLE tuvieron sustituciones de integrasa G/D/E193D y G193G/E emergentes del tratamiento en la semana 84 y la semana 108, respectivamente, y 1 sujeto con 275 copias por ml de ARN del VIH-1 tuvo un Q157Q/P emergente del tratamiento con sustitución de integrasa detectada en la semana 24. Ninguno de estos sujetos tuvo una disminución correspondiente en la susceptibilidad a dolutegravir. No se observó resistencia genotípica emergente del tratamiento al régimen de fondo en el grupo de dolutegravir en los ensayos SPRING-2 o SINGLE. No se observaron sustituciones de resistencia primaria emergentes del tratamiento en ninguno de los grupos de tratamiento en el ensayo FLAMINGO hasta la semana 96. Sujetos con tratamiento previo e inhibidores de la transferencia de integrasa sin tratamiento previo: En el grupo de dolutegravir del ensayo SAILING para sujetos con tratamiento previo, sin experiencia en INSTI (n=354), se observaron sustituciones de integrasa emergentes del tratamiento en 6 de 28 (21 %) sujetos que tenían datos de falla virológica y resistencia. En 5 de los 6 aislamientos, las sustituciones de INSTI emergentes incluyeron L74L/M/I, Q95Q/L, V151V/I (n=1 cada una) y R263K (n=2). El cambio en la susceptibilidad fenotípica de dolutegravir para estos 5 sujetos aislados fue menos de 2 veces. Un sujeto aislado tenía sustituciones de resistencia a raltegravir preexistentes E138A, G140S y Q148H al inicio del estudio y sustituciones emergentes de resistencia a INSTI T97A y E138A/T con reducción correspondiente de 148 veces en la susceptibilidad de dolutegravir al fracaso. En el grupo comparador de raltegravir, 21 de 49 (43 %) sujetos con datos de resistencia post basal tenían evidencia de sustituciones emergentes de resistencia a INSTI (L74M, E92Q, T97A, E138Q, G140S/A, Y143R/C, Q148H/R, Vl51I, Nl55H, El57Q y G163K/R) y resistencia enotípica a raltegravir. Sujetos virológicamente suprimidos: SWORD-1 y SWORD-2 son ensayos idénticos en sujetos virológicamente suprimidos que recibieron 2 NRTI más un INSTI, un NNRTI o un IP, que cambiaron a dolutegravir más rilpivirina (n=513) o permanecieron con su régimen antiviral actual (n=511). Dos sujetos en cada grupo de tratamiento habían confirmado la falla virológica en cualquier momento hasta la semana 48. Los 2 sujetos en el grupo de dolutegravir/rilpivirina tenían sustituciones de resistencia detectables en el rebote. Un sujeto tenía la sustitución K101K/E asociada a resistencia a NNRTI sin disminución de la susceptibilidad a rilpivirina (cambio de plegado=1.2) en la semana 36, no tenía sustituciones asociadas a resistencia de INSTI o disminución de la susceptibilidad a dolutegravir (cambio de plegado menor a 2) y tenía ARN del VIH-1 <50 copias por ml en la visita de retiro. El otro sujeto tenía la sustitución G193E asociada a la resistencia a dolutegravir al inicio del estudio (mediante secuenciación exploratoria del archivo de ADN pro viral del VIH) y en la semana 24 (mediante secuenciación convencional) sin disminución de la susceptibilidad a dolutegravir (cambio de plegado=1.02) en la semana 24. No se observaron sustituciones asociadas a resistencia en los otros dos sujetos en el grupo de régimen antirretroviral actual comparativo. Sujetos con tratamiento previo y experiencia con inhibidores de la transferencia de cadena de integrasa: En VIKING-3 se examinó la eficacia de dolutegravir 50 mg, 2 veces al día, más terapia de fondo optimizada, en sujetos con falla virológica previa o actual en un régimen que contiene INSTI- (elvitegravir o raltegravir). El uso de dolutegravir en pacientes con experiencia en INSTI debe guiarse por el número y tipo de sustituciones iniciales de INSTI. La eficacia de dolutegravir 50 mg, 2 veces al día, se reduce en pacientes con una sustitución de resistencia a INSTI Q148 más 2 o más sustituciones de resistencia a INSTI adicionales, que incluyen T66A, L741/M, E138A/K/T, G140S/A/C, Y143R/C/H, E157Q, G163S/E/K/Q o G193E/R. Respuesta por genotipo basal: De los 183 sujetos con datos basales, el 30 % albergaba virus con una sustitución en Q148 y el 33 % no tenía sustituciones primarias de resistencia a INSTI (T66A/I/K, E92Q/V, Y143R/C/H, Q148H/R/K y N155H) al inicio del estudio, pero tenían evidencia genotípica histórica de sustituciones de resistencia a INSTI, evidencia fenotípica de resistencia a elvitegravir o raltegravir, o evidencia genotípica de sustituciones de resistencia a INSTI en la selección. Las tasas de respuesta por genotipo basal se analizaron en un análisis «tal como se trató» en la semana 48 (n=175) (tabla 1). La tasa de respuesta en la semana 48 a los regímenes que contienen dolutegravir fue del 47 % (24 de 51) cuando las sustituciones Q148 estaban presentes al inicio del estudio; Q148 siempre estuvo presente con sustituciones adicionales de resistencia a INSTI (tabla 1). Además, se observó una respuesta virológica disminuida del 40 % (6 de 15) cuando la sustitución E157Q o K estaba presente al inicio con otras sustituciones de resistencia a INSTI, pero sin sustitución Q148H o R.
Respuesta por fenotipo basal: Las tasas de respuesta por fenotipo basal se estudiaron en un análisis de tratamiento con todos los sujetos con fenotipo basal disponibles hasta la semana 48 (n=163) (tabla 2). Estos grupos fenotípicos se basan en sujetos inscritos en VIKING-3 y no están destinados a representar puntos de corte de susceptibilidad clínica definitivos para dolutegravir. Los datos se proporcionan para guiar a los médicos sobre la probabilidad de éxito virológico, basado en la susceptibilidad al tratamiento previo a dolutegravir en pacientes resistentes a INSTI.
Resistencia al tratamiento con inhibidor de la transferencia de cadena de integrasa: Hubo 50 sujetos con falla virológica en el régimen de dolutegravir 2 veces al día en VIKING-3 con ARN del VIH-1 >400 copias por ml en el punto de falla, en la semana 48 o más, o en el último punto temporal del ensayo. Treinta y nueve sujetos con insuficiencia virológica tenían datos de resistencia que se utilizaron en el análisis de la semana 48. En el análisis de resistencia de la semana 48, el 85 % (33 de 39) de los sujetos con fracaso virológico tuvieron sustituciones resistentes a INSTI emergentes del tratamiento en sus aislamientos. La sustitución de resistencia a INSTI emergente de tratamiento más común fue T97A. Otras sustituciones de resistencia a INSTI frecuentemente emergentes incluyeron L74M, I o V, E138K o A, G140S, Q148H, Ro K, M154I o N155H. Las sustituciones E92Q, Y143R o C/H, S147G, V151A y E157E/Q emergieron en aislamientos de 1 a 3 sujetos. Al fracaso, la mediana del cambio de pliegue de dolutegravir de la referencia fue de 61 veces (rango: 0.75 a 209) para los aislamientos con sustituciones emergentes de resistencia a INSTI (n=33). La resistencia a 1 o más fármacos de fondo en el régimen de dolutegravir 2 veces al día también surgió en el 49 % (19 de 39) de sujetos en el análisis de resistencia de la semana 48. En VIKING-4 (ING116529), 30 sujetos con falla virológica actual en un régimen que contenía INSTI y evidencia genotípica de sustituciones de resistencia a INSTI en la selección fueron aleatorizados para recibir dolutegravir 50 mg, 2 veces al día, o placebo con el régimen de falla actual para 7 días y luego todos los sujetos recibieron dolutegravir/régimen abierto más un régimen de fondo optimizado a partir del día 8. Las respuestas virológicas en la semana 48 por categorías de Resistencia a INSTI, genotípicas y fenotípicas iniciales y las sustituciones asociadas a resistencia a INSTI que surgieron en el tratamiento con dolutegravir en VIKING-4, fueron consistentes con las observadas en VIKING-3. Cepas de VIH-1 y mutantes resistentes a inhibidores de la transferencia de cadena de integrasa, dirigidas al sitio de resistencia cruzada: La susceptibilidad de dolutegravir se probó contra 60 virus VIH-1 mutantes dirigidos al sitio INSTI-resistente (28 con sustituciones únicas y 32 con 2 o más sustituciones) y 6 virus VIH-2 mutantes dirigidos al sitio INSTI-resistente. Las sustituciones únicas de resistencia a INSTI T66K, I151L y S153Y confieren una disminución de más de 2 veces en la susceptibilidad a dolutegravir (rango: 2.3 a 3.6 veces la de la referencia). Las combinaciones de sustituciones múltiples T66K/L74M, E92Q/N155H, G140C/Q148R, G140S/Q148H, Ro K, Q148R/N155H, T97A/G140S/Q148 y las sustituciones en E138/G140/Q148, mostraron una disminución de más de 2 veces la susceptibilidad a dolutegravir (rango: 2.5 a 21 veces la de la referencia). En los mutantes del VIH-2, las combinaciones de sustituciones Al53G/Nl55H/S163G y E92Q/T97A/N155H/S163D confieren disminuciones de 4 veces en la susceptibilidad a dolutegravir, y E92Q / N155H y G140S / Q148R mostraron una disminución de 8.5 y 17 veces en la sensibilidad a dolutegravir, respectivamente. Cepas resistentes al inhibidor de la transcriptasa inversa y al inhibidor de la proteasa: Dolutegravir demostró una actividad antiviral equivalente contra 2 clones mutantes de VIH-1 resistentes a NNRTI, 3 resistentes a NRTI y 2 resistentes a PI, en comparación con la cepa de tipo salvaje. Farmacocinética: Bioequivalencia: Santigrase ha demostrado equivalencia terapéutica. Las propiedades farmacocinéticas de dolutegravir se han evaluado en sujetos adultos sanos y sujetos adultos infectados con VIH-1. La exposición a dolutegravir fue generalmente similar entre sujetos sanos y sujetos infectados por VIH-1. La exposición no lineal de dolutegravir después una dosificación de 50 mg, 2 veces al día, en comparación con 50 mg, 1 vez al día, en sujetos infectados con VIH-1 (tabla 3), se atribuyó al uso de inductores metabólicos en los regímenes antirretrovirales de fondo de sujetos que recibieron dolutegravir 50 mg, 2 veces al día, en ensayos clínicos.
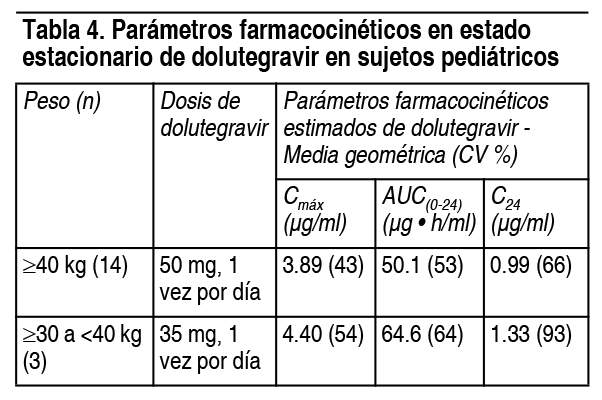
Absorción: Después de la administración oral de dolutegravir, se observaron concentraciones plasmáticas máximas 2 a 3 horas posteriores a la ingesta del fármaco. Con la dosificación 1 vez al día, el estado de equilibrio farmacocinético se logra en, aproximadamente, 5 días, con relaciones de acumulación promedio para AUC, Cmáx y C24 h que varían de 1.2 a 1.5. Las concentraciones plasmáticas de dolutegravir aumentaron de manera menos que proporcional a la dosis por encima de 50 mg. Dolutegravir es un sustrato de P-gp in vitro. No se ha establecido la biodisponibilidad absoluta de dolutegravir. Efecto de los alimentos: Dolutegravir puede tomarse con o sin alimentos. Los alimentos aumentaron el grado y disminuyeron la velocidad de absorción de dolutegravir. Las comidas bajas, moderadas y altas en grasa aumentaron el AUC(0-∞) de dolutegravir en un 33 %, 41 % y 66 %, respectivamente; la Cmáx aumentó en un 46 %, 52 % y 67 %, respectivamente y el Tmáx prolongado a 3, 4 y 5 horas desde 2 horas en ayunas, respectivamente. Distribución: Dolutegravir está altamente unido (≥98.9 %) a las proteínas plasmáticas humanas según los datos in vivo y la unión es independiente de la concentración plasmática del fármaco. El volumen aparente de distribución (Vd/F) posadministración de 50 mg, 1 vez al día, se estima en 17.4 l, según un análisis farmacocinético de la población. Líquido cefalorraquídeo (LCR): En 12 sujetos sin tratamiento previo, la dosis de dolutegravir 50 mg, diarios, más abacavir/lamivudina, produjo una concentración media de dolutegravir en LCR de 13.2 ng por ml (rango: 3.74 ng/ml a 18.3 ng/ml) de 2 a 6 horas posdosis, después de 16 semanas de tratamiento. La relevancia clínica de este hallazgo no se ha establecido. Eliminación: Dolutegravir tiene una vida media terminal de, aproximadamente, 14 horas y un aclaramiento aparente (CL/F) de 1.0 l por hora, según los análisis farmacocinéticos de la población. Metabolismo: Dolutegravir se metaboliza principalmente a través de UGT1A1 con alguna contribución de CYP3A. Polimorfismos en enzimas metabolizadoras de fármacos: En un metaanálisis de ensayos de sujetos sanos, los pacientes con genotipos UGT1A1 (n=7) que confieren un metabolismo pobre de dolutegravir tuvieron un aclaramiento 32 % menor de dolutegravir y un AUC 46 % mayor, en comparación con sujetos con genotipos asociados con metabolismo normal a través de UGT1A1 (n=41). Excreción: Después de una dosis oral única de 14C-dolutegravir, el 53 % de la dosis oral total se excretó sin cambios en las heces. El 31 % de la dosis oral total se excretó en la orina, representada por un glucurónido de éter de dolutegravir (18.9 % de la dosis total), un metabolito formado por oxidación en el carbono bencílico (3.0 % de la dosis total) y su producto hidrolítico N-desalquilado (3.6 % de la dosis total). La eliminación renal del fármaco inalterado fue baja (menos del 1 % de la dosis). Poblaciones específicas: Pacientes pediátricos: La farmacocinética de dolutegravir en nińos infectados con VIH-1 (n=17), que pesan al menos 30 kg (dosificados por bandas de peso, que reciben 35 mg o 50 mg), fueron similares a los observados en adultos infectados con VIH-1 que recibieron 50 mg de dolutegravir, 1 vez al día (tabla 4).
Pacientes geriátricos: El análisis farmacocinético de la población indicó que la edad no tuvo un efecto clínicamente relevante sobre la farmacocinética de dolutegravir. Pacientes con insuficiencia hepática: En un ensayo que comparó 8 sujetos con insuficiencia hepática moderada (puntuación B de Child-Pugh), con 8 controles sanos pareados, la exposición de dolutegravir a partir de una dosis única de 50 mg fue similar entre los 2 grupos. No se ha estudiado el efecto de la insuficiencia hepática grave (puntuación C de Child-Pugh) sobre la farmacocinética de dolutegravir. Pacientes con insuficiencia renal: En un ensayo que comparó a 8 sujetos con insuficiencia renal grave (CrCL menos de 30 ml por minuto), con 8 controles sanos emparejados, el AUC, la Cmáx y la C24 de dolutegravir fueron inferiores en un 40 %, 23 % y 43 %, respectivamente, en comparación con los valores de sujetos sanos pareados. El análisis farmacocinético de los datos de la población en los ensayos SAILING y VIKING-3 indicó que la insuficiencia renal leve y moderada no tuvo un efecto clínicamente relevante sobre la exposición a dolutegravir. Dolutegravir no se ha estudiado en pacientes que requieren diálisis. Pacientes coinfectados por VHB o VHC: Los análisis de población que utilizan datos farmacocinéticos agrupados de ensayos en adultos indicaron que no existe un efecto clínicamente relevante de la coinfección por VHC en la farmacocinética de dolutegravir. Los datos sobre la coinfección por VHB fueron limitados. Sexo y raza: Los análisis de población que utilizaron datos farmacocinéticos agrupados de ensayos en adultos indicaron que el sexo y la raza no tuvieron un efecto clínicamente relevante sobre la exposición a dolutegravir.
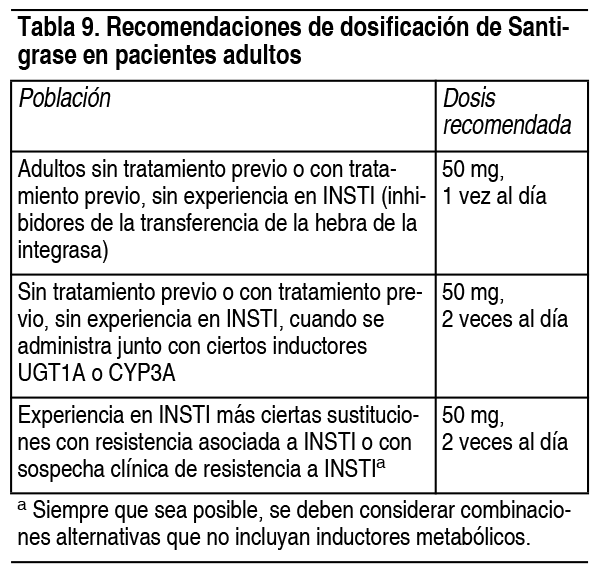
Posología:Adultos: Los comprimidos de Santigrase pueden tomarse con o sin alimentos.
Pacientes pediátricos: Los comprimidos de Santigrase pueden tomarse con o sin alimentos. Sin tratamiento previo o con tratamiento previo, sin experiencia en INSTI. La dosis recomendada de Santigrase, en pacientes pediátricos que pesen 40 kg o más, se proporciona en la tabla 2.
No se ha establecido la seguridad y eficacia de dolutegravir en pacientes pediátricos con experiencia en INSTI y con resistencia documentada o sospechada clínicamente a otros INSTI (raltegravir, elvitegravir).
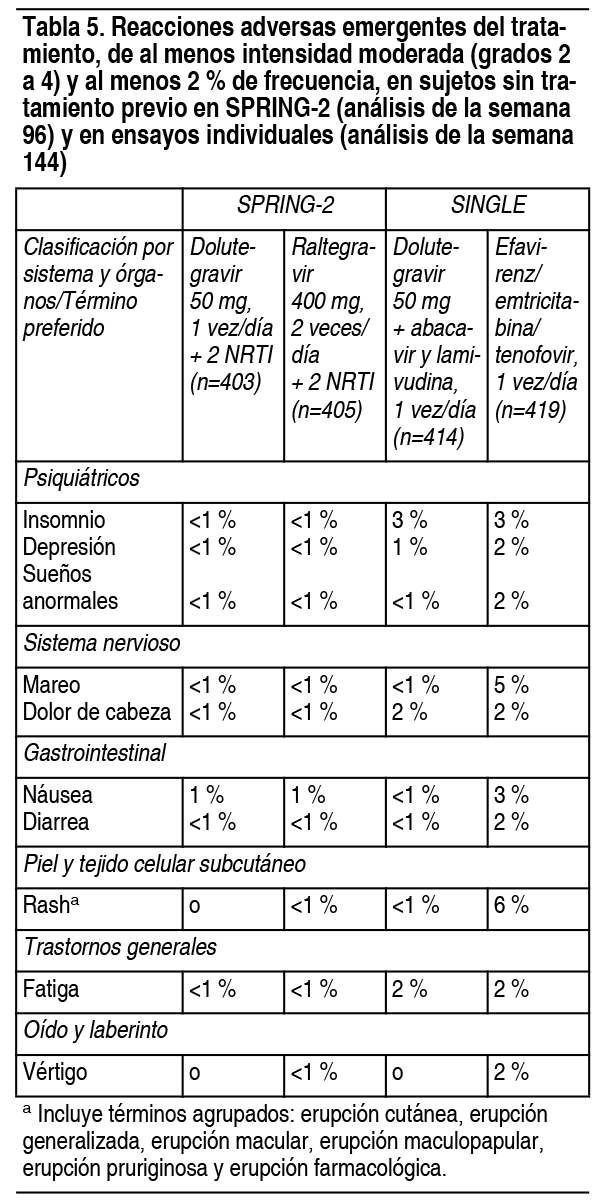
Efectos Colaterales:Experiencia en ensayos clínicos: Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los diversos estudios de un medicamento no se pueden comparar directamente con las tasas en los ensayos clínicos de otro medicamento y pueden no reflejar las tasas observadas en la práctica. Ensayos clínicos en adultos sin tratamiento previo: La evaluación de seguridad de dolutegravir en sujetos infectados con VIH-1 sin tratamiento, se basa en el análisis de datos de 2 estudios multicéntricos internacionales doble ciego, SPRING-2 (ING113086) y SINGLE (ING114467) y datos del ensayo internacional, multicéntrico y abierto FLAMINGO (ING114915). En SPRING-2, se asignaron 822 sujetos al azar que recibieron al menos 1 dosis de dolutegravir 50 mg, 1 vez al día, o raltegravir 400 mg, 2 veces al día, ambos en combinación con un tratamiento de dosis fija de inhibidor de la transcriptasa inversa de nucleósido dual (NRTI) (sulfato de abacavir y lamivudina o emtricitabina/tenofovir). Hubo 808 sujetos incluidos en los análisis de eficacia y seguridad. Durante 96 semanas, la tasa de eventos adversos que condujeron a la interrupción fue del 2 % en ambos grupos de tratamiento. En SINGLE, se asignaron 833 sujetos al azar que recibieron al menos 1 dosis de dolutegravir 50 mg con sulfato de abacavir y lamivudina en dosis fija, 1 vez al día, o efavirenz/emtricitabina/tenofovir en dosis fija, 1 vez al día (el tratamiento del estudio fue ciego hasta la semana 96 y abierto desde la semana 96 hasta la semana 144). Durante 144 semanas, las tasas de eventos adversos que condujeron a la interrupción fueron del 4 % en sujetos que recibieron dolutegravir 50 mg, 1 vez al día, más sulfato de abacavir y lamivudina y del 14 % en sujetos que recibieron efavirenz/emtricitabina/tenofovir, 1 vez al día. Las reacciones adversas (AR) emergentes del tratamiento de intensidad moderada a severa observadas en al menos el 2 % de los sujetos en cualquier grupo de tratamiento en los ensayos SPRING-2 y SINGLE se proporcionan en la tabla 5. La tabulación lado a lado es para simplificar la presentación. No se deben hacer comparaciones directas entre los ensayos debido a sus diferentes diseńos.
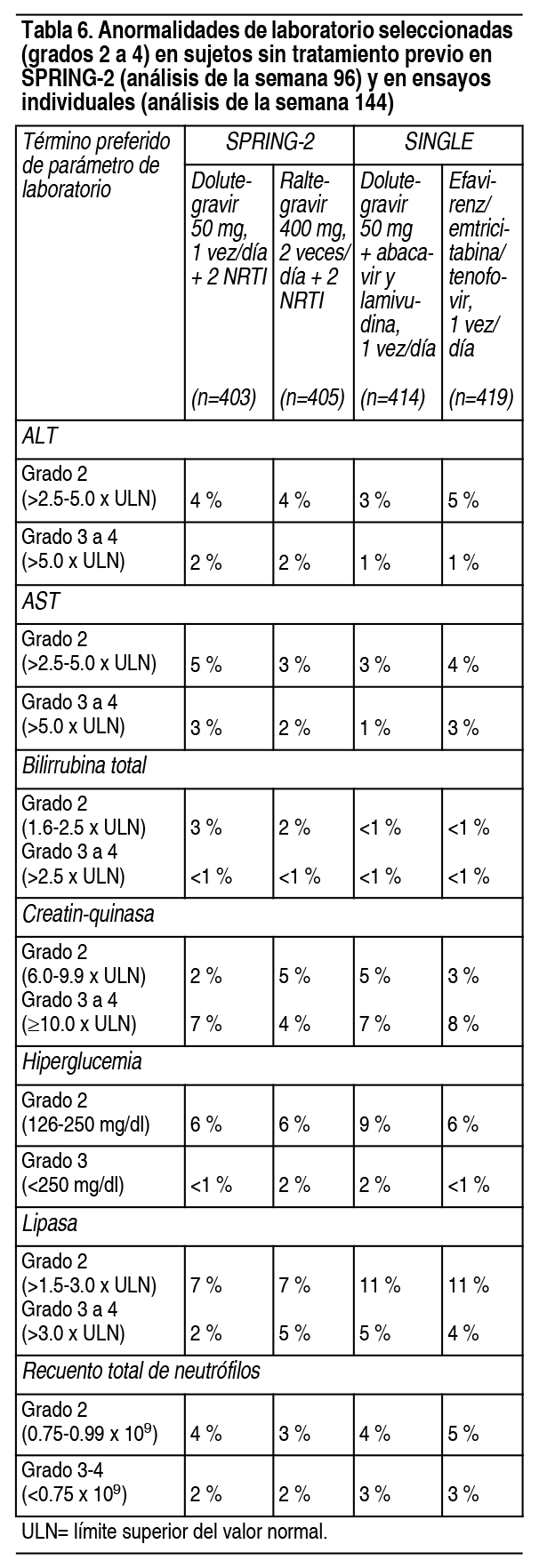
Por otra parte, el 1 % y menos del 1 % de los sujetos que recibieron dolutegravir y raltegravir, respectivamente, en SPRING-2 informaron insomnio de grado 1; en SINGLE las tasas fueron de 7 % y 4 % para dolutegravir y efavirenz/emtricitabina/tenofovir, respectivamente. Estos eventos no fueron limitantes del tratamiento. En un ensayo multicéntrico abierto (FLAMINGO), 243 sujetos recibieron 50 mg de dolutegravir, 1 vez al día, vs. 242 sujetos que recibieron darunavir 800 mg/ritonavir 100 mg, 1 vez al día, ambos en combinación con el régimen de fondo NRTI seleccionado por el investigador (abacavir y lamivudina o efavirenz/emtricitabina/tenofovir). Hubo 484 sujetos incluidos en los análisis de eficacia y seguridad. A lo largo de 96 semanas, las tasas de eventos adversos que condujeron a la interrupción fueron del 3 % en sujetos que recibieron dolutegravir y del 6 % en sujetos que recibieron darunavir/ritonavir. Las AR observadas en FLAMINGO fueron generalmente consistentes con las observadas en SPRING-2 y SINGLE. Sujetos con experiencia en tratamiento, no tratados con inhibidores de la transferencia de integrasa: En un ensayo multicéntrico internacional, doble ciego (ING111762, SAILING), 719 adultos infectados con VIH-1 con experiencia en tratamiento antirretroviral fueron asignados al azar y recibieron 50 mg de dolutegravir, 1 vez al día, o raltegravir 400 mg, 2 veces al día, con un régimen de fondo, seleccionado por el investigador, que constaba de hasta 2 agentes, incluido al menos uno completamente activo. A las 48 semanas, las tasas de AR que condujeron a la suspensión fueron del 3 % en sujetos que recibieron dolutegravir 50 mg, 1 vez al día + régimen de fondo y del 4 % en sujetos que recibieron raltegravir 400 mg, 2 veces al día + régimen de fondo. La única AR emergente del tratamiento, de intensidad moderada a severa con al menos 2 % de frecuencia en cualquiera de los grupos de tratamiento, fue la diarrea: 2 % (6 de 354) en sujetos que recibieron dolutegravir 50 mg, 1 vez al día + régimen de fondo y 1 % (5 de 361) en sujetos que recibieron raltegravir 400 mg, 2 veces al día + régimen de fondo. Sujetos con experiencia en el tratamiento y experimentados con inhibidores de la transferencia de integrasa: En un ensayo multicéntrico, abierto, de 1 solo grupo (ING112574, VIKING-3), 183 adultos infectados por VIH, con experiencia en tratamiento antirretroviral, con insuficiencia virológica y con evidencia actual o histórica de resistencia a raltegravir o elvitegravir recibieron 50 mg de dolutegravir, 2 veces al día, más el régimen de fondo deficiente actual durante 7 días y una terapia de fondo optimizada desde el día 8. La tasa de eventos adversos que llevaron a la interrupción fue del 4 % de los sujetos en la semana 48. Las AR emergentes del tratamiento en VIKING-3 fueron generalmente similares, en comparación con las observaciones con la dosis de 50 mg, 1 vez al día, en ensayos de Fase 3 para adultos. Sujetos virológicamente suprimidos: Las AR observadas para dolutegravir más rilpivirina en el análisis de la semana 48 sobre datos agrupados de 2 ensayos idénticos, internacionales, multicéntricos, abiertos (SWORD-1 y SWORD-2) de 513 sujetos infectados por VIH-1, suprimidos virológicamente, que cambiaron de su régimen antirretroviral actual a dolutegravir más rilpivirina, fueron consistentes con los perfiles de AR y la gravedad de los componentes individuales, cuando se les administraron otros agentes antirretrovirales. No hubo AR (grados 2 a 4) con una incidencia de al menos 2 % en ninguno de los grupos de tratamiento. Las tasas de eventos adversos que llevaron a la interrupción fueron del 4 % en los sujetos que recibieron dolutegravir más rilpivirina, 1 vez al día, y menos del 1 % en los sujetos que permanecieron en su régimen antirretroviral actual. Reacciones adversas menos comunes observadas en ensayos con pacientes sin tratamiento previo y con experiencia en el tratamiento: Las siguientes AR ocurrieron en menos del 2 % de los sujetos sin tratamiento o con experiencia en tratamiento, que recibieron dolutegravir en un régimen combinado en cualquier ensayo. Estos eventos se han incluido debido a su seriedad y evaluación de la posible relación causal. Trastornos gastrointestinales: Dolor abdominal, molestias abdominales, flatulencia, dolor abdominal superior, vómitos. Trastornos hepatobiliares: Hepatitis. Trastornos musculoesqueléticos: Miositis. Trastornos psiquiátricos: Ideación suicida, intento de suicidio, comportamiento suicida o consumación. Estos eventos se observaron principalmente en sujetos con antecedentes de depresión u otras enfermedades psiquiátricas. Trastornos renales y urinarios: Insuficiencia renal. Trastornos de la piel y del tejido subcutáneo: Prurito. Anormalidades de laboratorio: Sujetos sin tratamiento previo: En la tabla 6 se presentan anormalidades de laboratorio seleccionadas (grados 2 a 4) con un grado de empeoramiento con respecto al valor inicial y que representan la toxicidad de peor grado en al menos el 2 % de los sujetos. El cambio medio respecto al valor inicial observado para los valores de lípidos seleccionados se presenta en la tabla 7. La tabulación lado a lado es para simplificar la presentación. No se deben hacer comparaciones directas entre los ensayos debido a poseen diferentes diseńos.
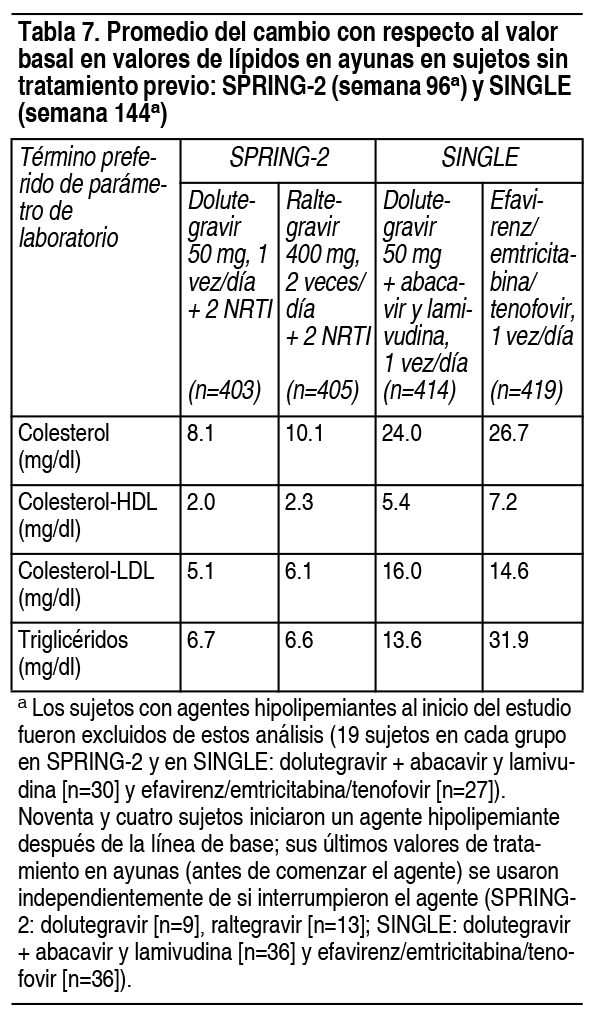
Las anomalías de laboratorio observadas en el ensayo FLAMINGO fueron generalmente consistentes con las observaciones en SPRING-2 y SINGLE. Sujetos tratados previamente, sin experiencia con inhibidores de la transferencia de integrasa: Las anomalías de laboratorio observadas en SAILING fueron generalmente similares, en comparación con las observadas en los ensayos sin tratamiento previo (SPRING-2 y SINGLE). Sujetos con tratamiento previo, experimentados con inhibidores de la transferencia de integrasa: Las anormalidades de laboratorio más comunes (mayores del 5 % para los grados 2 a 4 combinados) observadas en VIKING-3 en la semana 48 fueron: elevación de ALT (9 %), AST (8 %), colesterol (10 %) y creatin-quinasa (6 %); hiperglicemia (14 %) y elevación de lipasa (10 %). Dos por ciento (4 de 183) de los sujetos tenían una anormalidad de laboratorio de hematología emergente del tratamiento de grados 3 a 4, siendo la neutropenia (2 % [3 de 183]) la más frecuentemente reportada. Adultos virológicamente suprimidos: Las anomalías de laboratorio observadas en SWORD-1 y SWORD-2 fueron generalmente similares, en comparación con las observadas en los otros ensayos de Fase 3. Coinfección por el virus de la hepatitis B y/o hepatitis C: En los ensayos de Fase 3, los sujetos con coinfección por el virus de la hepatitis B y/o C pudieron inscribirse, siempre que las pruebas de química del hígado basales no superaran 5 veces el límite superior de lo normal (ULN). En general, el perfil de seguridad en sujetos con coinfección por virus de hepatitis B y/o C fue similar al observado en sujetos sin coinfección por hepatitis B o C, aunque las tasas de AST y ALT anormales fueron más altas en el subgrupo con hepatitis B y/o C para todos los grupos de tratamiento. Se observaron anormalidades de ALT de grados 2 a 4 en sujetos coinfectados con hepatitis B y/o C, en comparación con sujetos con VIH monoinfectados que recibieron dolutegravir, en 18 % frente a 3 %, con la dosis de 50 mg, 1 vez al día y 13 % frente a 8 %, con la dosis de 50 mg, 2 veces al día. Se observaron elevaciones de las enzimas hepáticas, compatibles con el síndrome de reconstitución inmune, en algunos sujetos con hepatitis B y/o C al comienzo de la terapia con dolutegravir, particularmente en el entorno donde se retiró la terapia contra la hepatitis. Cambios en la creatinina sérica: Se ha demostrado que dolutegravir aumenta la creatinina sérica, debido a la inhibición de la secreción tubular de creatinina, sin afectar la función glomerular renal. Se produjeron aumentos en la creatinina sérica dentro de las primeras 4 semanas de tratamiento, permaneciendo estable durante 96 semanas. En sujetos sin tratamiento previo, se observó un cambio medio desde el inicio de 0.15 mg/dl (rango: 0.32 mg/dl a 0.65 mg/dl), después de 96 semanas de tratamiento. Los aumentos de creatinina fueron comparables con los NRTI de fondo y fueron similares en sujetos con tratamiento. Experiencia de ensayos clínicos en la población pediátrica: IMPAACT P1093 es un ensayo continuo, multicéntrico, abierto y no comparativo de, aproximadamente, 160 sujetos pediátricos, de 4 semanas de edad a menos de 18 ańos, infectados con VIH-1, de los cuales 46 (de 6 ańos a menos de 18 ańos) contaban con tratamiento previo, sin experiencia con INSTI. El perfil de reacción adversa fue similar al de los adultos. Las AR de grado 2 informadas por más de un sujeto fueron disminución del recuento de neutrófilos (n=3) y diarrea (n=2). No se informaron AR de grados 3 o 4 relacionadas con las drogas en estudio. Ninguna AR condujo a la interrupción del tratamiento. Las anormalidades de laboratorio de grados 3 o 4, informadas en más de un sujeto, fueron elevación de la bilirrubina total (n=3) y disminución del recuento de neutrófilos (n=2). Los cambios en la creatinina sérica media fueron similares a los observados en adultos. Experiencia posterior a la comercialización: Además de las reacciones adversas informadas en los ensayos clínicos, se han identificado los siguientes eventos durante el uso posterior a la comercialización. Debido a que estas reacciones se informan voluntariamente y se originan en una población de tamańo incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco. Trastornos hepatobiliares: Insuficiencia hepática aguda, hepatotoxicidad. Trastornos musculoesqueléticos: Mialgia. Trastornos psiquiátricos: Ansiedad.
Contraindicaciones:Santigrase está contraindicado en pacientes: Con reacción de hipersensibilidad previa a dolutegravir. Que reciben dofetilida, debido al potencial de aumento de las concentraciones plasmáticas de dofetilida y al riesgo de eventos graves o potencialmente mortales.
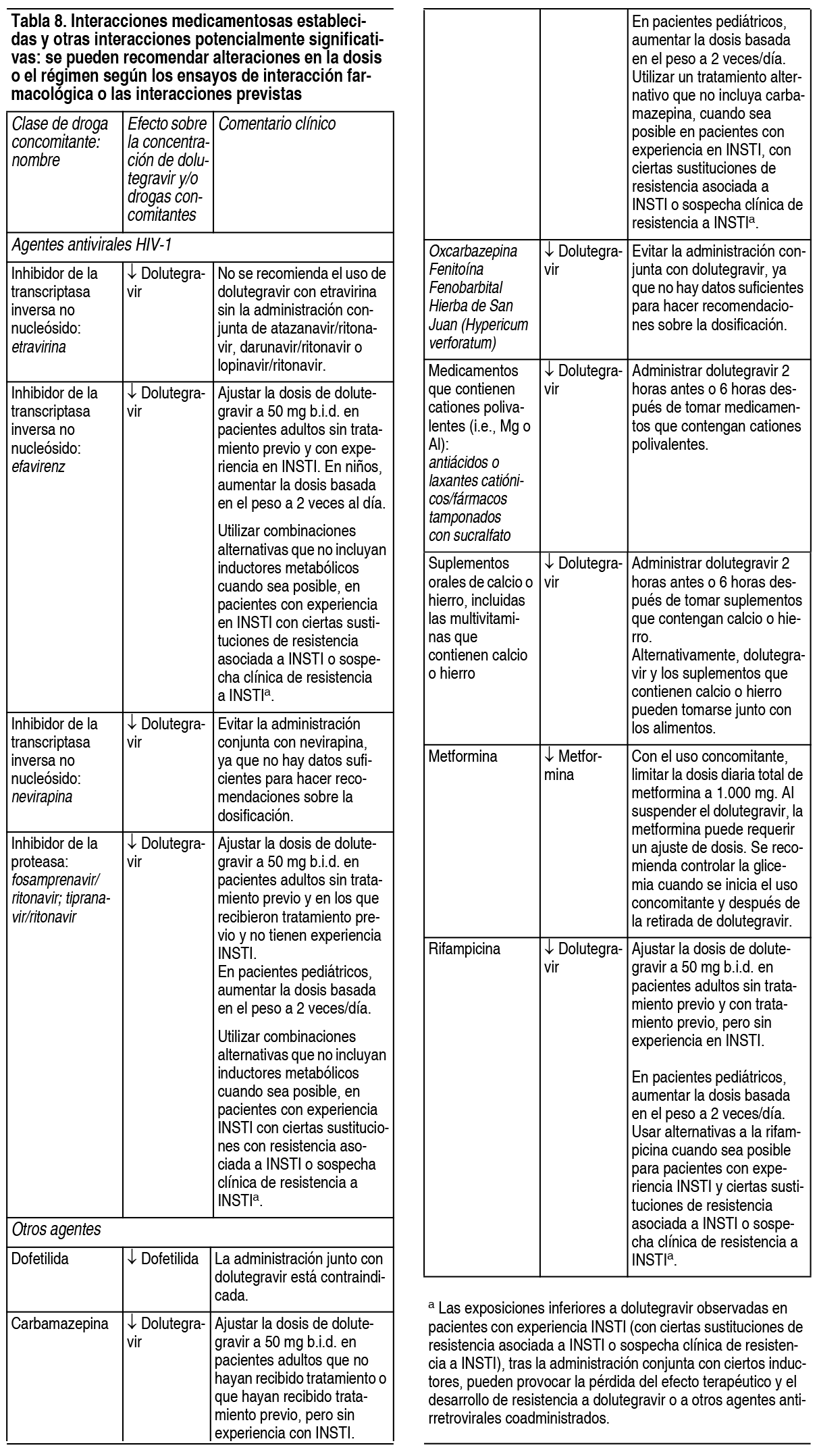
Advertencias:Reacciones de hipersensibilidad: Se notificaron reacciones de hipersensibilidad caracterizadas por erupción cutánea, hallazgos constitucionales y, a veces, disfunción orgánica, incluida la lesión hepática. Los eventos se informaron en menos del 1 % de los sujetos que recibieron dolutegravir en los ensayos clínicos de Fase 3. Suspender inmediatamente el dolutegravir y otros agentes sospechosos si se desarrollan signos o síntomas de reacciones de hipersensibilidad (que incluyen, entre otros, erupción cutánea grave o erupción cutánea acompańada de fiebre, malestar general, fatiga, dolores musculares o articulares, ampollas o descamación de la piel, ampollas orales o lesiones, conjuntivitis, edema facial, hepatitis, eosinofilia, angioedema, dificultad para respirar). Se debe controlar el estado clínico, incluidas las aminotransferasas hepáticas, y se debe iniciar la terapia adecuada. El retraso en la interrupción del tratamiento con dolutegravir u otros agentes sospechosos después del inicio de la hipersensibilidad puede provocar una reacción potencialmente mortal. Dolutegravir está contraindicado en pacientes que han experimentado una reacción de hipersensibilidad previa al fármaco. Hepatotoxicidad: Se han informado eventos adversos hepáticos en pacientes que reciben dolutegravir. Los pacientes con hepatitis B o C subyacente pueden tener un mayor riesgo de empeoramiento o desarrollo de elevaciones de transaminasas con el uso del fármaco. En algunos casos, las elevaciones en las transaminasas fueron consistentes con el síndrome de reconstitución inmune o la reactivación de la hepatitis B, particularmente en el entorno donde se retiró la terapia contra la hepatitis. Se han notificado casos de toxicidad hepática que incluyen elevación de enzimas hepáticas, hepatitis e insuficiencia hepática aguda en pacientes que reciben un régimen que contiene dolutegravir sin enfermedad hepática preexistente u otros factores de riesgo identificables. En protocolos con abacavir, dolutegravir y lamivudina se han informado lesiones hepáticas inducidas por fármacos, que conducen a un trasplante de hígado. Se recomienda monitorizar la hepatotoxicidad. Riesgo de reacciones adversas o pérdida de la respuesta virológica debido a interacciones farmacológicas: El uso concomitante de dolutegravir y otras drogas puede dar lugar a interacciones farmacológicas conocidas o potencialmente significativas, algunas de las cuales pueden provocar: Pérdida del efecto terapéutico de dolutegravir y posible desarrollo de resistencia. Posibles reacciones adversas clínicamente significativas de mayores exposiciones de fármacos concomitantes. Para medicamentos concomitantes a los que se puede mitigar la interacción, consultar la tabla 3: varios pasos para prevenir o controlar estas interacciones medicamentosas significativas posibles y conocidas, incluidas las recomendaciones de dosificación. Considerar el potencial de interacciones farmacológicas antes y durante la terapia con dolutegravir; revisar los medicamentos concomitantes durante la terapia con el fármaco; monitorear las reacciones adversas asociadas con las drogas concomitantes. Síndrome de reconstitución inmunitaria: Se ha notificado el síndrome de reconstitución inmunitaria en pacientes tratados con terapia antirretroviral combinada, incluido dolutegravir. Durante la fase inicial del tratamiento antirretroviral combinado, los pacientes cuyos sistemas inmunes responden pueden desarrollar una respuesta inflamatoria a infecciones oportunistas indolentes o residuales (como infección por Mycobacterium avium, citomegalovirus, neumonía por Pneumocystis jirovecii [PCP] o tuberculosis), lo que puede requerir una evaluación adicional y tratamiento. También se ha informado que los trastornos autoinmunes (como la enfermedad de Graves, la polimiositis y el síndrome de Guillain-Barré) ocurren en el contexto de la reconstitución inmune; sin embargo, el tiempo de inicio es más variable y puede ocurrir muchos meses después del inicio del tratamiento.
Precauciones:Fertilidad, embarazo y lactancia: Embarazo: Resumen de riesgos: No hay datos suficientes en humanos sobre el uso de dolutegravir durante el embarazo para informar un riesgo de defectos de nacimiento y aborto espontáneo. Dado el número limitado de embarazos expuestos a regímenes basados en dolutegravir informados a la APR, no se pueden sacar conclusiones definitivas sobre la seguridad del fármaco en el embarazo. En los estudios de reproducción en animales no se observó evidencia de resultados adversos en el desarrollo con dolutegravir. Durante la organogénesis en ratas y conejos, las exposiciones sistémicas (AUC) a dolutegravir fueron menores en conejos que la exposición en humanos y, aproximadamente, 27 veces mayores en ratas a la dosis humana máxima recomendada (MRHD). En el estudio de desarrollo pre- y posnatal en ratas, la exposición sistémica materna (AUC) a dolutegravir fue, aproximadamente, 27 veces la exposición en humanos con la MRHD. Lactancia: Resumen de riesgo: Los Centros para el Control y la Prevención de Enfermedades recomiendan que las madres infectadas con VIH-1 en los Estados Unidos no amamanten a sus bebés, para evitar el riesgo de transmisión posnatal de la infección por VIH-1. No se sabe si dolutegravir está presente en la leche materna humana, si afecta la producción de leche humana o si tiene efectos en el lactante. Cuando se administró a ratas lactantes, dolutegravir estaba presente en la leche. Debido al potencial de (1) transmisión del VIH-1 (en lactantes sin VIH) y (2) desarrollo de resistencia viral (en lactantes con VIH), se recomienda indicar a las madres que no amamanten si están recibiendo dolutegravir. Efectos sobre la capacidad para conducir y utilizar maquinaria: Se debe informar a los pacientes que se han notificado mareos durante el tratamiento con dolutegravir. El estado clínico del paciente y el perfil de reacciones adversas de dolutegravir deben tenerse en cuenta al considerar la capacidad del paciente para conducir vehículos u operar maquinaria.
Interacciones Medicamentosas:Efecto de dolutegravir sobre la farmacocinética de otros agentes:In vitro, dolutegravir inhibió los transportadores catiónicos orgánicos renales, OCT2 (IC50=1.93 µM) y el transportador de extrusión de toxinas y múltiples fármacos (MATE1 (IC50=6.34 µM). In vivo, dolutegravir inhibe la secreción tubular de creatinina al inhibir OCT2 y potencialmente MATE1. Dolutegravir puede aumentar las concentraciones plasmáticas de los fármacos eliminados mediante OCT2 o MATE1, dofetilida y metformina (tabla 8). Dolutegravir inhibió in vitro los transportadores renales basolaterales, el transportador de aniones orgánicos (OAT1) (IC50=2.12 µM) y OAT3 (IC50=1.97 µM). Sin embargo, in vivo, dolutegravir no alteró las concentraciones plasmáticas de tenofovir o para-aminohipurato, sustratos de OAT1 y OAT3. In vitro, dolutegravir no inhibió (IC50 >50 µM) lo siguiente: citocromo P450 (CYP) 1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A, uridina difosfato (UDP)-glucuronosil transferasa 1A1 (UGT1A1), UGT2B7, glicoproteína P (P-gp), proteína de resistencia al cáncer de mama (BCRP), bomba de exportación de sales biliares (BSEP), polipéptido transportador de aniones orgánicos (OATP)1B1, OATP1B3, OCT1, proteína de resistencia a múltiples fármacos (MRP)2 o MRP4. In vitro, dolutegravir no indujo CYP1A2, CYP2B6 o CYP3A4. Según estos datos y los resultados de los ensayos de interacción farmacológica, no se espera que dolutegravir afecte la farmacocinética de los medicamentos que son sustratos de estas enzimas o transportadores. Efecto de otros agentes sobre la farmacocinética de dolutegravir: Dolutegravir es metabolizado por UGT1A1 con alguna contribución de CYP3A. Dolutegravir también es un sustrato de UGT1A3, UGT1A9, BCRP y P-gp in vitro. Los medicamentos que inducen esas enzimas y transportadores pueden disminuir la concentración plasmática y reducir el efecto terapéutico de dolutegravir. La administración conjunta de dolutegravir y otras drogas que inhiben estas enzimas puede aumentar la concentración plasmática de dolutegravir. La etravirina redujo significativamente las concentraciones plasmáticas de dolutegravir, pero el efecto de la etravirina se mitigó mediante la administración conjunta de lopinavir/ritonavir o darunavir/ritonavir y se espera que mitigue atazanavir/ritonavir (tabla 8). In vitro, dolutegravir no resultó ser un sustrato de OATP1B1 o OATP1B3. Interacciones establecidas y otras interacciones potencialmente significativas: La tabla 8 proporciona recomendaciones clínicas como resultado de las interacciones farmacológicas con dolutegravir. Estas recomendaciones se basan en ensayos de interacción farmacológica o interacciones predichas debido a la magnitud esperada de la interacción y el potencial de eventos adversos graves o pérdida de eficacia.
Fármacos sin interacciones clínicamente significativas con dolutegravir: Según los resultados de los ensayos de interacciones farmacológicas, los siguientes fármacos pueden coadministrarse con dolutegravir sin un ajuste de dosis: Atazanavir/ritonavir, darunavir/ritonavir, daclatasvir, elbasvir/grazoprevir, metadona, midazolam, omeprazol, anticonceptivos orales que contienen norgestimato y etinilestradiol, prednisona, rifabutina, rilpivirina, sofosbuvir/velpatasvir y tenofovir.
Sobredosificación: No se conoce un tratamiento específico para la sobredosis con dolutegravir. Si se produce una sobredosificación, se debe controlar al paciente y aplicar el tratamiento de soporte estándar según sea necesario. Dado que dolutegravir está altamente unido a las proteínas plasmáticas, es poco probable que se elimine significativamente por diálisis.
 Laboratorio
Laboratorio