Composición: Cada comprimido bucodispersable contiene: 10 mg de Bilastina. Excipientes c.s.: Manitol, Croscarmelosa Sódica, Fumarato de Estearilo y Sodio, Sucralosa, Aroma de Uva Roja. Forma farmacéutica: Comprimido bucodispersable. Comprimidos redondos, ligeramente biconvexos y blancos de 8 mm de diámetro.
Acción Terapéutica:Grupo farmacoterapéutico: Antihistamínicos de uso sistémico, otros antihistamínicos de uso sistémico. Código ATC: R06AX29.
Indicaciones: Tratamiento sintomático de la rinoconjuntivitis alérgica (estacional y perenne) y de la urticaria. Blaxitec ODT 10 mg comprimidos bucodispersables está indicado en nińos de 2 a 11 ańos.
Propiedades:Propiedades farmacológicas: Mecanismo de acción: Bilastina es un antagonista de la histamina no sedante y de acción prolongada, con afinidad antagonista selectiva por los receptores H1 periféricos y sin afinidad por los receptores muscarínicos. Tras la administración de una dosis única, bilastina inhibió durante 24 horas las reacciones cutáneas de habón y eritema inducidas por histamina. Propiedades farmacocinéticas: Absorción: Bilastina se absorbe rápidamente tras la administración oral, con un tiempo de, aproximadamente, 1.3 horas, hasta alcanzar la concentración plasmática máxima. No se ha observado acumulación. La biodisponibilidad oral media de bilastina es del 61 %. Distribución: Estudios in vitro e in vivo han demostrado que bilastina es un substrato de la P-gp (ver la sección «Interacción con ketoconazol o eritromicina» e «Interacción con diltiazem») y del OATP (ver la sección «Interacción con zumo de pomelo»). A las dosis terapéuticas, la unión de bilastina a las proteínas plasmáticas es de 84-90 %. Biotransformación: En estudios in vitro, bilastina no indujo ni inhibió la actividad de los isoenzimas del CYP450. Eliminación: En un estudio de balance de masas, realizado en voluntarios adultos sanos, tras la administración de una dosis única de 20 mg de 14C-bilastina, casi el 95 % de la dosis administrada fue recuperada en orina (28.3 %) y heces (66.5 %) como bilastina inalterada, confirmando que bilastina no es significativamente metabolizada en humanos. La vida media de eliminación calculada en voluntarios sanos fue de 14.5 horas. Linealidad: Bilastina presenta una farmacocinética lineal en el rango de dosis estudiado (5 a 220 mg), con una baja variabilidad interindividual. Insuficiencia renal: Los efectos de bilastina, en pacientes con insuficiencia renal, han sido estudiados en adultos. En un estudio realizado en sujetos con insuficiencia renal, la AUC0-∞ media (DE) aumentó de 737.4 (±260.8) ngxhr/ml, en sujetos sin insuficiencia (IFG: >80 ml/min/1.73 m2), a 967.4 (±140.2) ngxhr/ml, en sujetos con insuficiencia leve (IFG: 50-80 ml/min/1.73 m2), 1384.2 (±263,23) ngxhr/ml, en sujetos con insuficiencia moderada (IFG: 30 - <50 ml/min/1.73 m2), y 1708.5 (±699.0) ngxhr/ml, en sujetos con insuficiencia grave (IFG: <30 ml/min/1.73 m2). La semivida de eliminación media (±DE) de bilastina fue de 9.3 horas (±2.8), en sujetos sin insuficiencia, 15.1 horas (±7.7), en sujetos con insuficiencia leve, 10.5 horas (±2.3), en sujetos con insuficiencia moderada, y 18.4 horas (±11.4), en sujetos con insuficiencia grave. La excreción urinaria de bilastina fue esencialmente completa tras 48-72 horas en todos los sujetos. No cabe esperar que estos cambios farmacocinéticos tengan una influencia clínicamente relevante sobre la seguridad de bilastina, ya que los niveles plasmáticos de bilastina, en pacientes con insuficiencia renal, continúan estando dentro del rango de seguridad de bilastina. Insuficiencia hepática: No hay datos farmacocinéticos en sujetos con insuficiencia hepática. Bilastina no es metabolizada en humanos. Puesto que los resultados del estudio en insuficiencia renal indican que la vía renal es la vía principal responsable de la eliminación, cabe esperar que la excreción biliar solo esté implicada de forma marginal en la eliminación de bilastina. No se espera que los cambios en la función hepática tengan una influencia clínicamente relevante en la farmacocinética de bilastina. Población pediátrica: Los datos farmacocinéticos en nińos han sido obtenidos de un estudio farmacocinético de fase II, que incluía 31 nińos de edades comprendidas entre 4 y 11 ańos con rinoconjuntivitis alérgica o urticaria crónica, a los cuales se les administró una toma diaria de bilastina 10 mg comprimidos bucodispersables. El análisis farmacocinético de los datos de concentraciones plasmáticas mostró que la dosis pediátrica de bilastina 10 mg, 1 vez al día, da como resultado una exposición sistémica equivalente a la observada tras administrar una dosis de 20 mg en adultos y adolescentes, siendo el valor de AUC promedio de 1014 ngxhr/ml para nińos de 2 a 11 ańos. Estos resultados estuvieron muy por debajo del umbral de seguridad, basado en los datos de dosis de 80 mg, 1 vez al día, en adultos, de acuerdo con el perfil de seguridad del medicamento. Estos resultados confirmaron la elección de bilastina vía oral, 1 vez al día, como la dosis terapéutica recomendada para población pediátrica, en el rango de edad de 2 a 11 ańos, con un peso corporal mínimo de 20 kg. Datos preclínicos sobre seguridad: Los datos de los estudios no clínicos con bilastina no muestran riesgos especiales para los seres humanos, según los estudios convencionales de farmacología de seguridad, toxicidad a dosis repetidas, genotoxicidad y potencial carcinogénico. En los estudios de toxicidad para la reproducción, únicamente se observaron efectos de bilastina sobre el feto (pérdidas pre- y posimplantación, en ratas, y osificación incompleta de los huesos craneales, el esternón y los miembros, en conejos) a dosis tóxicas para la madre. Los niveles de exposición determinados por las NOAEL son superiores (>30 veces) a los niveles de exposición alcanzados en humanos a la dosis terapéutica recomendada. En un estudio sobre lactancia, se detectó bilastina en la leche de ratas en período de lactancia, tras la administración oral de una dosis única (20 mg/kg). Las concentraciones de bilastina en la leche fueron alrededor de la mitad de las del plasma materno. Se desconoce la relevancia de estos resultados para los humanos. En un estudio de fertilidad en ratas, la administración oral de bilastina a dosis hasta 1000 mg/kg/día no indujo ningún efecto sobre los órganos reproductivos de los machos ni de las hembras. Los índices de apareamiento, fertilidad y gravidez no se vieron afectados. Tal y como se observó en un estudio de distribución en ratas, con determinación de las concentraciones de fármaco por autorradiografía, bilastina no se acumula a nivel del SNC.
Posología:Población pediátrica: Nińos de 2 a 11 ańos: 10 mg de bilastina (1 comprimido bucodispersable), 1 vez al día, para el alivio de los síntomas de la rinoconjuntivitis alérgica (rinitis alérgica estacional y perenne) y de la urticaria. El comprimido bucodispersable debe administrarse 1 hora antes o 2 horas después de la ingesta de alimentos o de zumos de frutas (ver la sección Interacción con otros medicamentos y otras formas de interacción). Nińos menores de 2 ańos: Los datos actualmente disponibles se incluyen en las secciones Advertencias y precauciones especiales de empleo, Reacciones adversas, Propiedades farmacodinámicas y Propiedades farmacocinéticas. Sin embargo, no se puede hacer una recomendación posológica. Por lo tanto, no se debe usar bilastina en este grupo de edad. En adultos y adolescentes (mayores de 12 ańos), la administración apropiada es bilastina 20 mg en comprimidos. Duración del tratamiento: Para rinoconjuntivitis alérgica, el tratamiento debe limitarse al período de exposición a los alérgenos. Para rinitis alérgica estacional, el tratamiento puede interrumpirse cuando se hayan resuelto los síntomas y reiniciarse en caso de que estos reaparezcan. En rinitis alérgica perenne, se puede proponer al paciente el tratamiento continuado durante los períodos de exposición a los alérgenos. Para urticaria, la duración del tratamiento depende del tipo, duración y evolución de los síntomas. Poblaciones especiales: Insuficiencia renal: No se ha establecido la seguridad y eficacia de bilastina en nińos con insuficiencia renal. Los estudios realizados en grupos de adultos con un riesgo especial (pacientes con insuficiencia renal) indican que no se requiere ajustar la dosis de bilastina en adultos (ver la sección Propiedades farmacocinéticas). Insuficiencia hepática: No se ha establecido la seguridad y eficacia de bilastina en nińos con insuficiencia hepática. No hay experiencia clínica en pacientes adultos ni pediátricos con insuficiencia hepática. Sin embargo, dado que bilastina no se metaboliza y se elimina inalterada en orina y heces, no se espera que la insuficiencia hepática aumente la exposición sistémica por encima del margen de seguridad, en pacientes adultos. Por ello, no se requiere ajustar la dosis en pacientes adultos con insuficiencia hepática (ver la sección Propiedades farmacocinéticas). Forma de administración: Vía oral. El comprimido bucodispersable se debe colocar en la boca, donde se disuelve rápidamente en la saliva, pudiendo tragarse fácilmente. Alternativamente, el comprimido bucodispersable se puede dispersar en agua antes de la administración. No se debe utilizar zumo de pomelo o cualquier otro zumo de fruta para la dispersión (ver la sección Interacción con otros medicamentos y otras formas de interacción).
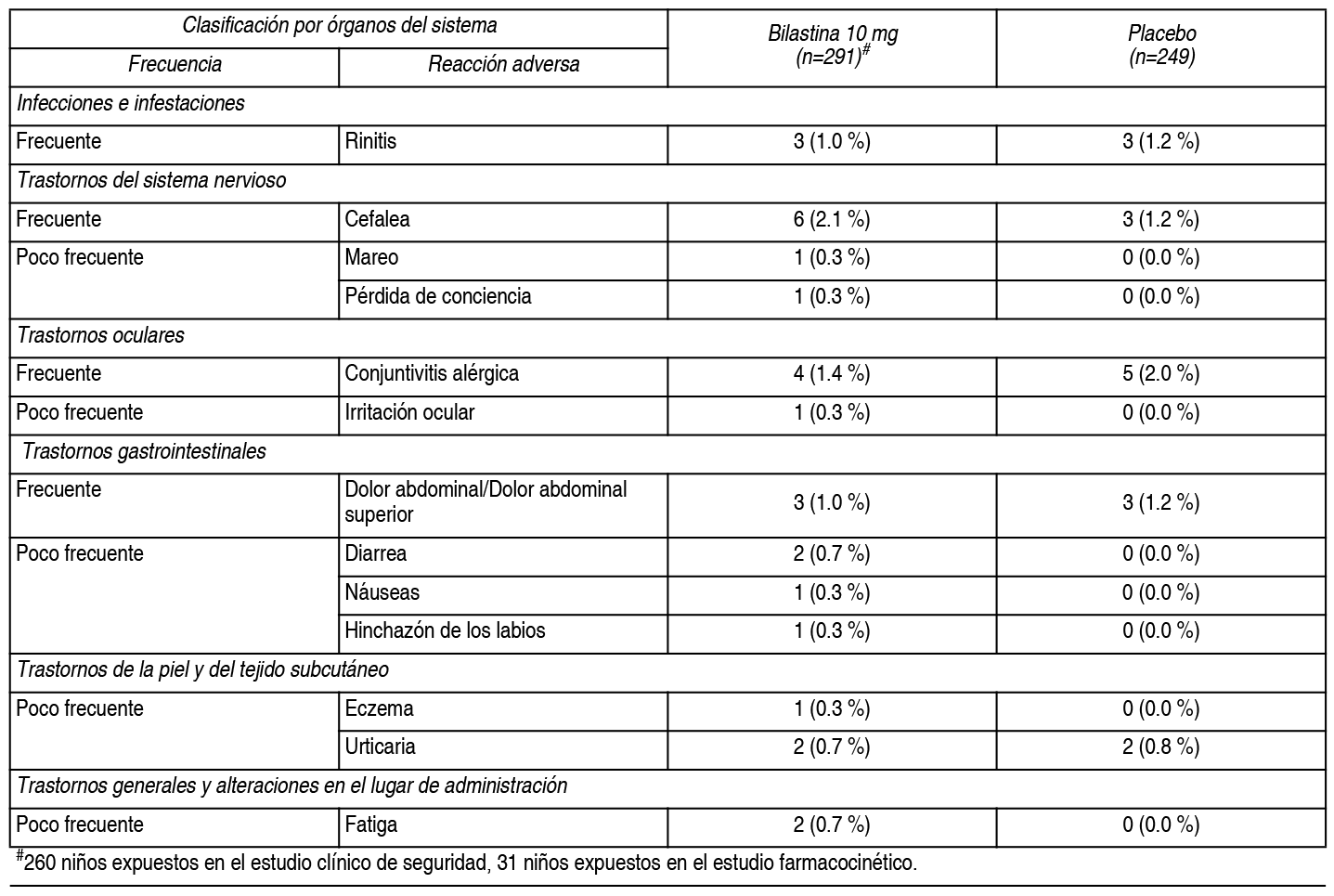
Efectos Colaterales:Resumen del perfil de seguridad en la población pediátrica: Durante el desarrollo clínico, la frecuencia, el tipo y la intensidad de las reacciones adversas en adolescentes (de 12 a 17 ańos), fueron las mismas que las observadas en adultos. La información recogida en esta población (adolescentes) durante la poscomercialización, ha confirmado los resultados de los ensayos clínicos. En un ensayo clínico controlado de 12 semanas, el porcentaje de nińos (2-11 ańos) que notificaron acontecimientos adversos (AA), después del tratamiento con bilastina 10 mg para rinoconjuntivitis alérgica o urticaria idiopática crónica, fue comparable con los pacientes que recibieron placebo (68.5 % vs. 67.5 %). Los AA relacionados notificados más frecuentemente por 291 nińos (2-11 ańos) que recibieron bilastina (en la forma farmacéutica de comprimidos bucodispersables), durante los ensayos clínicos (#260 nińos expuestos en el estudio clínico de seguridad, 31 nińos expuestos en el estudio farmacocinético), fueron dolor de cabeza, conjuntivitis alérgica, rinitis y dolor abdominal. Estos acontecimientos adversos relacionados, ocurrieron con una frecuencia comparable en 249 pacientes que recibieron placebo. Resumen tabulado de reacciones adversas en la población pediátrica: La siguiente tabla muestra los acontecimientos adversos al menos posiblemente relacionados con bilastina y notificados en más del 0.1 % de los nińos (2-11 ańos) tratados con bilastina, durante el desarrollo clínico. Las frecuencias se han clasificado de la siguiente forma: Muy frecuentes (≥1/10), frecuentes (≥1/100 a <1/10), poco frecuentes (≥1/1000 a <1/100), raras (≥1/10 000 a <1/1000), muy raras (<1/10 000) y frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones «raras», «muy raras» y de «frecuencia no conocida» no se han incluido en la tabla.
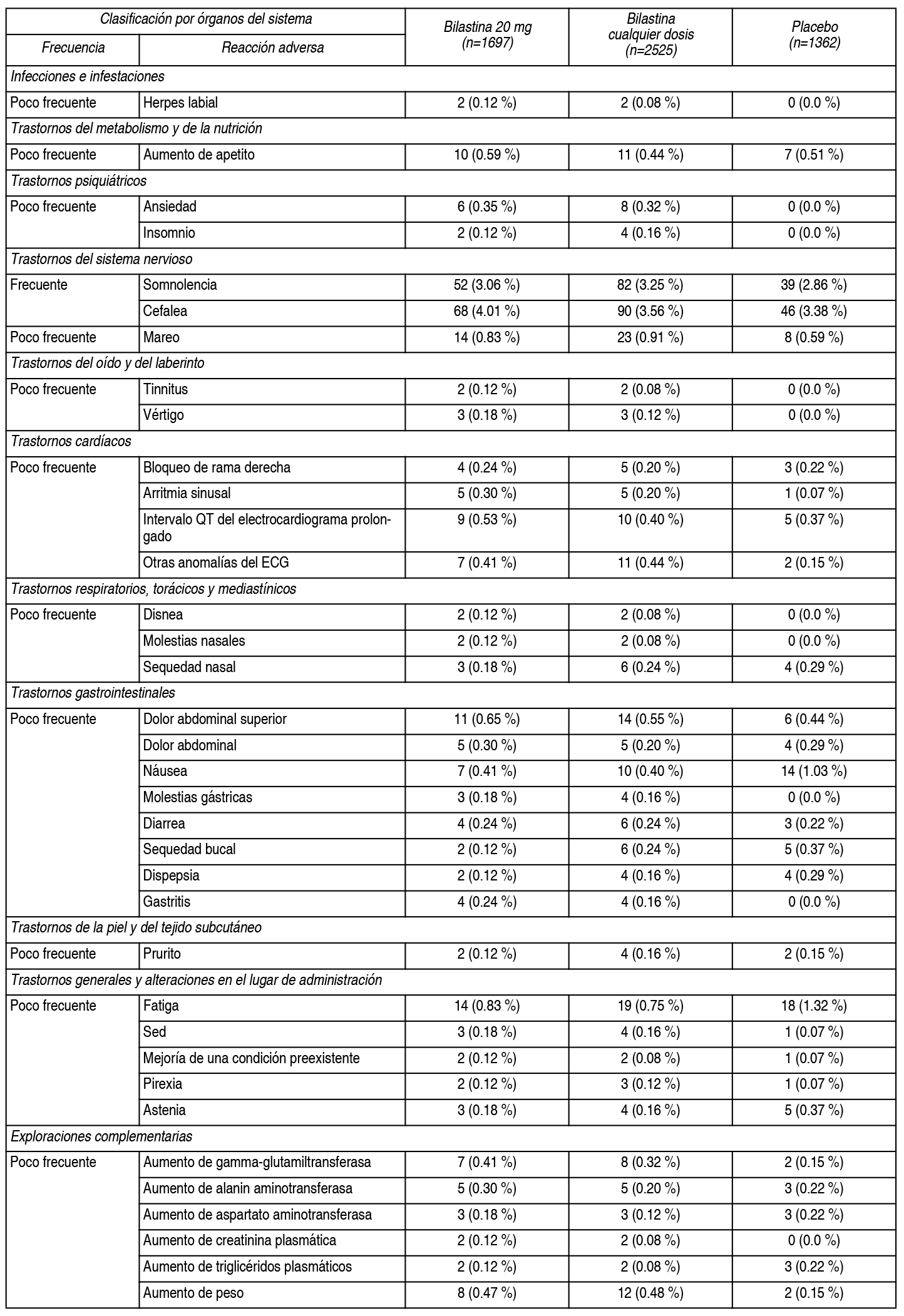
Descripción de las reacciones adversas relevantes en la población pediátrica: Se observaron cefalea, dolor abdominal, conjuntivitis alérgica y rinitis, tanto en nińos tratados con 10 mg de bilastina, como con placebo. La frecuencia notificada fue 2.1 % vs. 1.2 % para cefalea, 1.0 % vs. 1.2 % para dolor abdominal, 1.4 % vs. 2.0 % para conjuntivitis alérgica y 1.0 % vs. 1.2 % para rinitis. Resumen del perfil de seguridad en pacientes adultos y adolescentes: La incidencia de acontecimientos adversos, en pacientes adultos y adolescentes afectados de rinoconjuntivitis alérgica o urticaria crónica idiopática, tratados con 20 mg de bilastina en los estudios clínicos, fue comparable a la incidencia en pacientes que recibieron placebo (12.7 % frente a 12.8 %). Los ensayos clínicos de fase II y III, realizados durante el desarrollo clínico, incluyeron 2525 pacientes adultos y adolescentes tratados con diferentes dosis de bilastina, de los cuales 1697 recibieron 20 mg de bilastina. En estos ensayos, 1362 pacientes recibieron placebo. Los acontecimientos adversos notificados más frecuentemente por los pacientes tratados con bilastina 20 mg, para la indicación de rinoconjuntivitis alérgica o urticaria crónica idiopática, fueron cefalea, somnolencia, mareo y fatiga. Estos acontecimientos adversos ocurrieron con una frecuencia similar en los pacientes que recibieron placebo. Resumen tabulado de reacciones adversas en pacientes adultos y adolescentes: La siguiente tabla, muestra las reacciones adversas, al menos posiblemente relacionadas con bilastina y notificadas, en más del 0.1 % de los pacientes tratados con bilastina 20 mg, durante el desarrollo clínico (n=1697). Las frecuencias se han clasificado de la siguiente forma: Muy frecuentes (≥1/10), frecuentes (≥1/100 a <1/10), poco frecuentes (≥1/1000 a <1/100), raras (≥1/10 000 a <1/1000), muy raras (<1/10 000) y frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones «raras», «muy raras» y de «frecuencia no conocida», no se han incluido en la tabla.
Frecuencia no conocida (no puede estimarse a partir de los datos disponibles): Se han observado palpitaciones, taquicardia, reacciones de hipersensibilidad (como anafilaxia, angioedema, disnea, erupción cutánea, edema localizado/hinchazón local y eritema) y vómitos, durante el período de poscomercialización. Descripción de las reacciones adversas relevantes en pacientes adultos y adolescentes: Se observaron somnolencia, cefalea, mareo y fatiga tanto en pacientes tratados con 20 mg de bilastina o con placebo. La frecuencia notificada fue 3.06 % vs. 2.86 % para somnolencia, 4.01 % vs. 3.38 % para cefalea, 0.83 % vs. 0.59 % para mareo y 0.83 % vs. 1.32 % para fatiga. La información recogida durante la poscomercialización, ha confirmado el perfil de seguridad observado durante el desarrollo clínico. Notificación de sospechas de reacciones adversas: Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Nacional de Farmacovigilancia de medicamentos de Uso Humano.
Contraindicaciones: Hipersensibilidad al principio activo o a alguno de los excipientes.
Advertencias:Población pediátrica: La eficacia y seguridad de bilastina, en nińos menores de 2 ańos, no han sido establecidas y hay poca experiencia clínica en nińos de 2 a 5 ańos, por lo que bilastina no se debería utilizar en estos grupos de edad. En pacientes con insuficiencia renal moderada o severa, la administración concomitante de bilastina con inhibidores de la P-glicoproteína, tales como ketoconazol, eritromicina, ciclosporina, ritonavir o diltiazem, puede aumentar los niveles plasmáticos de bilastina y, por tanto, aumentar el riesgo de efectos adversos de bilastina. Por ello, la administración concomitante de bilastina e inhibidores de la P-glicoproteína, debe evitarse en pacientes con insuficiencia renal moderada o severa. Este medicamento contiene menos de 1 mmol de sodio (23 mg), por comprimido bucodispersable; esto es, esencialmente «exento de sodio».
Precauciones:Fertilidad, embarazo y lactancia: Embarazo: No hay datos relativos al uso de bilastina en mujeres embarazadas o estos son limitados. Los estudios en animales no sugieren efectos perjudiciales directos ni indirectos en términos de toxicidad para la reproducción, el parto o el desarrollo posnatal (ver la sección «Datos preclínicos sobre seguridad»). Como medida de precaución, es preferible evitar el uso de Blaxitec ODT durante el embarazo. Lactancia: La excreción de bilastina en la leche no ha sido estudiada en humanos. Los datos farmacocinéticos disponibles en animales, muestran que bilastina se excreta en la leche (ver la sección «Datos preclínicos sobre seguridad»). Se debe decidir si continuar/discontinuar la lactancia o interrumpir/abstenerse del tratamiento con Blaxitec ODT, tras considerar el beneficio de la lactancia para el nińo y el beneficio del tratamiento con bilastina para la madre. Fertilidad: No hay datos clínicos o estos son limitados. En un estudio en ratas no se detectó ningún efecto negativo sobre la fertilidad (ver la sección «Datos preclínicos sobre seguridad»). Efectos sobre la capacidad para conducir y utilizar máquinas: Un estudio realizado en adultos, para evaluar los efectos de bilastina sobre la capacidad para conducir, demostró que el tratamiento con 20 mg de bilastina no afectó al rendimiento durante la conducción. No obstante, dado que puede variar la respuesta individual al medicamento, se recomienda a los pacientes no conducir o utilizar máquinas hasta que hayan establecido su propia respuesta a bilastina.
Interacciones Medicamentosas:Interacción con otros medicamentos y otras formas de interacción: Los estudios de interacciones se han realizado solo en adultos y se resumen a continuación: Interacción con alimentos: Los alimentos reducen significativamente la biodisponibilidad oral de bilastina 20 mg comprimidos en un 30 % y de bilastina 10 mg comprimidos bucodispersables en un 20 %. Interacción con zumo de pomelo: La administración concomitante de bilastina 20 mg y zumo de pomelo, disminuyó la biodisponibilidad de bilastina en un 30 %. Este efecto puede ocurrir también con otros zumos de frutas. El grado de reducción en la biodisponibilidad puede variar entre fabricantes y frutos. El mecanismo responsable de esta interacción es la inhibición del OATP1A2, un transportador de captación, del cual bilastina es sustrato (ver la sección «Propiedades farmacocinéticas»). Los medicamentos que sean sustratos o inhibidores de OATP1A2, tales como ritonavir o rifampicina, podrían igualmente reducir las concentraciones plasmáticas de bilastina. Interacción con ketoconazol o eritromicina: La administración concomitante de bilastina 20 mg, 1 vez al día, y ketoconazol 400 mg, 1 vez al día, o eritromicina 500 mg, 3 veces al día, aumentó el AUC de bilastina en 2 veces y la Cmáx en 2-3 veces. Estos cambios se pueden explicar debido a la interacción con transportadores intestinales de excreción, ya que bilastina es sustrato de la P-gp y no es metabolizada (ver la sección «Propiedades farmacocinéticas»). Estos cambios no parecen afectar al perfil de seguridad de bilastina y ketoconazol o eritromicina, respectivamente. Otros medicamentos que sean sustratos o inhibidores de la P-gp, tal como ciclosporina, podrían igualmente aumentar las concentraciones plasmáticas de bilastina. Interacción con diltiazem: La administración concomitante de bilastina 20 mg, 1 vez al día, y diltiazem 60 mg, 1 vez al día, aumentó la Cmáx de bilastina un 50 %. Este efecto se puede explicar por la interacción con transportadores intestinales de excreción (ver la sección «Propiedades farmacocinéticas») y no parece afectar al perfil de seguridad de bilastina. Interacción con alcohol: El rendimiento psicomotor tras la administración concomitante de alcohol y 20 mg de bilastina, 1 vez al día, fue similar al observado tras la administración de alcohol y placebo. Interacción con lorazepam: La administración concomitante de bilastina 20 mg, 1 vez al día, y lorazepam 3 mg, 1 vez al día, durante 8 días, no potenció los efectos depresores del SNC de lorazepam. Población pediátrica: No se han realizado estudios de interacciones en nińos con los comprimidos bucodispersables de bilastina. Dado que no hay experiencia clínica sobre la interacción de bilastina con otros medicamentos, alimentos o zumos de frutas en nińos, actualmente se deben considerar los resultados obtenidos en los estudios de interacciones con adultos cuando se prescriba bilastina pediátrica. No existen datos clínicos en nińos para asegurar que los cambios en la AUC o Cmáx debido a interacciones, afectan al perfil de seguridad de bilastina.
Sobredosificación: No hay datos de sobredosis en nińos. La información relacionada con sobredosis aguda de bilastina, se recoge de la experiencia de los ensayos clínicos realizados durante el desarrollo en adultos y la vigilancia poscomercialización. En los ensayos clínicos, tras la administración de bilastina a dosis de 10 a 11 veces la dosis terapéutica (220 mg, como dosis única, o 200 mg/día, durante 7 días) a 26 voluntarios adultos sanos, la frecuencia de acontecimientos adversos tras el tratamiento fue 2 veces superior a la observada tras la administración de placebo. Las reacciones adversas más frecuentemente notificadas fueron mareo, cefalea y náusea. No se notificaron acontecimientos adversos graves ni prolongaciones significativas del intervalo QTc. La información recogida durante la poscomercialización, coincide con la información obtenida en los ensayos clínicos. La evaluación crítica del efecto de dosis múltiples de bilastina (100 mg, durante 4 días) sobre la repolarización ventricular en «un estudio cruzado de Thorough QT/QTc», realizado con 30 voluntarios adultos sanos, no mostró ninguna prolongación significativa del intervalo QTc. En caso de producirse una sobredosis, se recomienda tratamiento sintomático y de soporte. No se conoce ningún antídoto específico para bilastina.
Conservación:Periodo de validez: 60 meses. Condiciones de almacenamiento: No almacenar a más de 30 °C.
 Laboratorio
Laboratorio