Composición: Cada comprimido sublingual contiene: Ketorolaco Trometamol 10 mg. Excipientes c.s.: Manitol, Crospovidona, Estearato de Magnesio, Ácido Cítrico, Dióxido de Silicio Coloidal, Sacarina Sódica, Sucralosa, Saborizante Artificial de Pińa.
Acción Terapéutica:Grupo terapéutico: Derivados de ácido acético y sustancias relacionadas; antiinflamatorios y antirreumáticos no esteroidales. Código ATC: M01AB15.
Indicaciones: Este medicamento está indicado como manejo a corto plazo del dolor.
Propiedades: Este medicamento es un potente agente analgésico de la clase de medicamentos antiinflamatorios no esteroidales (AINE). No es un opiáceo y no presenta efectos sobre receptores opiáceos. Su mecanismo de acción es a través de inhibición del sistema enzimático ciclooxigenasa y, en consecuencia, de la síntesis de prostaglandinas. Puede ser considerado como un analgésico de actividad periférica. Su actividad biológica está asociada a su forma S. Este medicamento no presenta propiedades sedantes ni ansiolíticas. Absorción y Distribución: El ketorolaco trometamol se absorbe rápida y completamente después de la administración oral. A pH fisiológico, la sal de ketorolaco trometamol se disocia completamente en forma de ketorolaco aniónico. Su biodisponibilidad varía entre 0.81 y 1.00, lo que sugiere poco o ningún metabolismo presistémico, por lo que no existe interacción con enzima de inducción. El pico plasmático es de 0.8 mg L-1 y ocurre entre 30 a 60 minutos después de la administración de dosis orales de 10 y 30 mg, respectivamente. La Tmáx ocurre tarde en ancianos, en pacientes con enfermedad renal o hepática y después de la ingestión de alimentos. La concentración plasmática máxima aumenta linealmente con la dosis. La vida media del ketorolaco trometamol es muy similar para las diferentes vías de administración (IV, IM u oral), con una media de 5.4 horas y un rango de 4.5 a 5.6 horas. El nivel plasmático en estado estacionario es de 0.6 a 0.8 mg. L-1 (rango 0.2 - 1.7. L-1) y 1.3 - 1.5 mg. L-1 (rango 0.3 - 3.5 mg. L-1) después de 24 horas de administración de ketorolaco trometamol 15 o 30 mg respectivamente, cada 6 horas. En el plasma, el ketorolaco trometamol se une en más de 99 % a proteínas, preferentemente a albúmina. La distribución es rápida, pero gran parte del compuesto se retiene en el compartimiento vascular debido al bajo volumen de distribución 0.11 - 0.25 lkg-1, que se duplica en nińos de 4 a 8 ańos. Sin embargo, como en estos pacientes el «aclaramiento» también es mayor, no hay cambios en la vida media plasmática del fármaco. La penetración de la barrera hematoencefálica es pobre con apenas 0.2 % de la concentración plasmática y la proporción cerebro/plasma de solo 0.03. Estudios en animales demostraron que la relación renal/plasma es 1.5, pero que la proporción tejido/plasma es menor a 1.0, lo que indica que no hay acumulación tisular del fármaco. El ketorolaco trometamol atraviesa la placenta y entra a la circulación fetal, alcanzando niveles sanguíneos en el feto de 11.6 % (rango 4 - 25 %) en relación con los niveles sanguíneos maternos. Como consecuencia, se observa un efecto antiagregante de plaquetas en el recién nacido. El ketorolaco trometamol se excreta poco en la leche materna y su concentración no supera 7.9 μg.L- 1, en un régimen de 10 mg cada 6 horas. La proporción leche/plasma es inferior a 0.04. Metabolismo y Excreción: Aproximadamente, el 40 % de la dosis de ketorolaco trometamol se metaboliza preferiblemente por vía hepática. La principal vía de excreción es la urinaria, con más de 90 % del fármaco inalterado, además de metabolitos. Un pequeńo porcentaje de la dosis (10 %) se excreta en las heces. El aclaramiento plasmático total en voluntarios jóvenes y sanos fue de 0.35 a 0.55 mL.min-1.Kg-1, mientras que en pacientes con dańo renal y en ancianos, el aclaramiento se reduce. La vida media de eliminación en ancianos fue de 6 a 7 horas, en pacientes con dańo renal 9 a 10 horas y en pacientes con cirrosis hepática de 5.4 horas. Los cambios en la farmacocinética de ketorolaco trometamol son raros y no requieren ningún cambio en el régimen de dosificación. No hay evidencia de relación alguna entre el efecto terapéutico de ketorolaco y su concentración plasmática. Absorción oral: >95 %. Metabolismo presistémico: <10 %. Vida media plasmática (rango): 4.4-5.6 h. Vida media plasmática (media): 5.4 h. Volumen de distribución: 0.11-0.3. kg-1. Unión a proteína plasmática: 99.2 %.
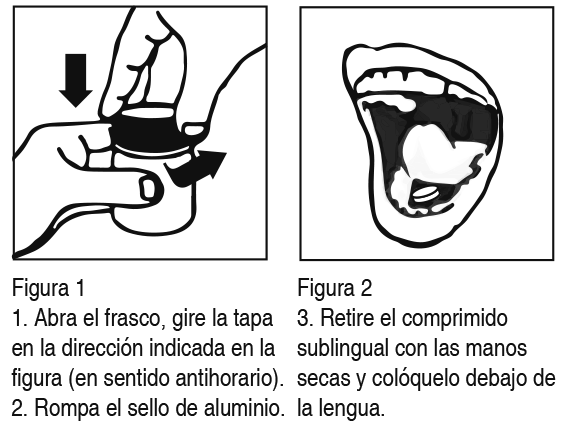
Posología: Los comprimidos sublinguales de ketorolaco trometamol deben ser colocados y mantenidos debajo de la lengua, hasta su completa disolución, de acuerdo con las siguientes orientaciones:
Retire el comprimido sublingual con las manos secas y colóquelo debajo de la lengua (Figura 2). Los frascos de ketorolaco trometamol contienen un agente secante de gel de sílice que no debe eliminarse ni ingerirse. Este medicamento no debe ser partido, abierto o masticado. Debido a que existe evidencia de efectos adversos relacionados con la dosis (sangramiento gastrointestinal), especialmente en pacientes geriátricos, deberá emplearse la menor dosis efectiva posible. Precaución especial, en cuanto a usar dosis reducidas, se deberá tener en cuenta para pacientes geriátricos, pacientes con un peso menor a 50 kg y en aquellos con insuficiencia renal. Para el manejo del dolor moderado a severo, se recomienda una dosis inicial de 20 mg (2 comprimidos) y posteriormente, dosis orales de 10 mg hasta 4 veces al día (a intervalos de 4-6 horas) las que son administradas según necesidad. Efectos adversos serios pueden presentarse con dosis orales que excedan los 40 mg diarios, sin mejorar la eficacia analgésica. Se recomienda solo en terapias cortas (hasta 14 días) y no se indica para el alivio del dolor crónico a largo plazo.
Efectos Colaterales:Los siguientes efectos adversos pueden ocurrir: Reacciones frecuentes (> 1/100 y <1/10): Dolor abdominal con cólicos, diarrea, mareos, somnolencia, dispepsia, edema, cefalea, náuseas. Reacciones poco frecuentes (> 1/1.000 y <1/100): Dermatitis alérgica, reacciones alérgicas, constipación, hiperhidrosis, hipertensión, aumento del apetito, flatulencia, prurito de piel, exantema cutáneo, estomatitis, urticaria y vómitos. Reacciones raras (>1/10.000 y <1/1.000): Úlcera péptica aguda con hemorragia y perforación, anafilaxia, anemia, anorexia, azotemia, sangrado de heridas, sangre en las heces, visión borrosa, asma bronquial, enfermedad pulmonar, tos, depresión, disgeusia, disnea, eosinofilia, epistaxis, eructos, euforia, dermatitis exfoliativa, enfermedad extrapiramidal, desmayo, fiebre, calofríos, dolor de garganta persistente, flatulencia, gastritis, debilidad general, alucinaciones, pérdida de audición, hematuria, hepatitis, agitación, aumento de frecuencia urinaria, infección, ictericia, edema laríngeo, nefritis, nerviosismo, oliguria, palidez, palpitaciones, parestesia, úlcera péptica, inhibición de la agregación plaquetaria, polidipsia, poliuria, proteinuria, edema pulmonar, púrpura, sangrado rectal, insuficiencia renal, rinitis, síndrome de Stevens-Johnson, trombocitopenia, zumbido, hinchazón de la lengua, temblores, retención urinaria, urticaria, mareos, aumento de peso, xerostomía, pruebas de función hepática anormales, accidente cerebrovascular, hepatitis por medicamentos, cólico renal, hemorragia y perforación gastrointestinal, infarto de miocardio, fácil aparición de hematomas/hemorragia, dificultad para respirar.
Contraindicaciones: Este medicamento está contraindicado para uso en pacientes con úlcera péptica (lesión del estómago o duodeno); sangrado gastrointestinal; sangrado cerebrovascular; diátesis hemorrágica (hemofilia), trastornos de coagulación sanguínea; postoperatorio de cirugía de revascularización miocárdica, con uso de anticoagulantes, incluyendo heparina en dosis bajas (2500-5000 unidades cada 12 horas); en postoperatorio con un alto riesgo de hemorragia u homeostasis incompleta; hipersensibilidad a ketorolaco trometamol, o a cualquiera de los componentes de la fórmula o a otros AINE (fármacos antiinflamatorios no esteroidales), en pacientes en los que el ácido acetilsalicílico o los inhibidores de síntesis de prostaglandinas inducen reacciones alérgicas; poliposis nasal y asma bronquial concomitantes, debido al riesgo de presentar reacción alérgica intensa (se han observado reacciones anafilácticas severas en estos pacientes); tratamiento concomitante con otros AINE, pentoxifilina, probenecid o sales de litio; hipovolemia o deshidratación; insuficiencia renal grave o moderada (creatinina sérica > 160 micromol/l); historia de asma; insuficiencia cardíaca crónica; enfermedad del sistema cardiovascular; evento de riesgo cardiovascular aumentado; infarto de miocardio; fumadores; colitis ulcerosa (úlceras de colon); embarazo, parto o lactancia. Este medicamento también está contraindicado en administración neuroaxial (epidural o intratecal) debido a la presencia de alcohol y como profiláctico en analgesia antes y durante la cirugía debido a inhibición de agregación plaquetaria y consiguiente aumento del riesgo de sangrado. Hipersensibilidad: Debido a la potencial hipersensibilidad cruzada con otros AINE, no deben administrarse a pacientes que han sufrido síntomas de asma, rinitis, urticaria, pólipos nasales, angioedema, broncoespasmo y otros síntomas o reacciones alérgicas o anafilactoides asociadas a ácido acetilsalicílico u otro AINE. En raros casos, se han presentado reacciones anafilácticas fatales y asmáticas severas. No debe usarse AINE con excepción del ácido acetilsalicílico en pacientes en el período posoperatorio inmediato a una cirugía de by pass coronario. Categoría de riesgo C: Este medicamento no se debe utilizar en mujeres embarazadas sin orientación médica o del cirujano dentista. Informe inmediatamente a su médico en caso de sospecha de embarazo.
Advertencias: Los médicos deben saber que el alivio del dolor en algunos pacientes puede no ocurrir dentro de los 30 minutos posteriores a la administración de este medicamento. Síntomas de toxicidad gastrointestinal severa tales como inflamación, sangramiento, ulceración y perforación del intestino grueso y delgado pueden ocurrir en cualquier momento con o sin síntomas previos, en pacientes en terapia crónica con AINE, por lo que se debe estar alerta frente a la presencia de síntomas de ulceración o sangrado. Se han producido reacciones anafilactoides en pacientes asmáticos, sin exposición previa a AINE, pero que han experimentado previamente rinitis con o sin pólipos nasales o que exhiben broncoespasmo potencialmente fatal después de tomar ácido acetilsalicílico u otro AINE. Debido al riesgo de que se produzcan eventos cardiovasculares severos con el uso de AINE, a excepción de ácido acetilsalicílico, debe evaluarse cuidadosamente la condición del paciente antes de prescribir estos medicamentos. Efectuar seguimiento de los pacientes en tratamiento crónico con AINE por signos y síntomas de ulceración o sangramiento del tracto gastrointestinal. Efectuar monitoreo de transaminasas y enzimas hepáticas en pacientes en tratamiento con AINE, especialmente en aquellos tratados con nimesulida, sulindaco, diclofenaco y naproxeno. Usar con precaución en pacientes con compromiso de la función cardíaca, hipertensión, terapia diurética crónica y otras condiciones que predisponen a retención de fluidos debido a que los AINE pueden causar retención de fluidos además de edema periférico. Se puede producir insuficiencia renal aguda, nefritis intersticial con hematuria, síndrome nefrótico, proteinuria, hiperkalemia, hiponatremia, necrosis papilar renal y otros cambios medulares renales. Pacientes con falla renal preexistente están en mayor riesgo de sufrir insuficiencia renal aguda. Una descompensación renal se puede precipitar en pacientes en tratamiento por AINE debido a una reducción de dosis dependiente en la formación de prostaglandinas afectando principalmente a ancianos, lactantes, prematuros, pacientes con falla renal, cardíaca o disfunción hepática, glomerulonefritis crónica, deshidratación, diabetes mellitus, septicemia, pielonefritis y depleción de volumen extracelular en aquellos que están tomando inhibidores de la ECA y/o diuréticos. Efectos gastrointestinales: Este medicamento puede causar irritación gastrointestinal, úlceras o sangrado en pacientes con o sin historia de síntomas previos, como con todos los AINE. Los pacientes ancianos y debilitados son más propensos a desarrollar estas reacciones. La incidencia aumenta con la dosis y la duración del tratamiento. En un estudio de vigilancia poscomercialización, no aleatorio, en hospital, se notificó un aumento del riesgo de sangrado gastrointestinal, clínicamente serio, en pacientes menores de 65 ańos que recibieron una dosis media superior a 90 mg de ketorolaco trometamol por vía intramuscular/intravenosa en comparación con pacientes que recibieron opiáceos por vía parenteral. Efectos respiratorios: El broncoespasmo puede precipitarse en pacientes con historia de asma. Efectos renales: Los medicamentos que inhiben la biosíntesis de prostaglandinas (incluidos los AINE) presentaron informes de nefrotoxicidad, incluyendo nefritis glomerular, nefritis intersticial, necrosis papilar renal, síndrome nefrótico e insuficiencia renal aguda. Se debe tener precaución en pacientes con insuficiencia renal o hepática, ya que el uso de AINE puede provocar deterioro de función renal. Después de una dosis de ketorolaco trometamol, se han notificado elevaciones de urea sérica, creatinina y potasio, al igual que con otros fármacos que inhiben la síntesis de prostaglandinas. Pacientes con insuficiencia renal: Dado que ketorolaco trometamol y sus metabolitos se excretan principalmente por los rińones, los pacientes con insuficiencia renal moderada a grave (creatinina sérica mayor a 160 micromol/l) no deben recibir este medicamento. Los pacientes con menos dańo renal deben recibir una dosis reducida de ketorolaco trometamol (que no exceda 60 mg/día intramuscular o intravenosa y que no exceda 40 mg/días comprimidos sublinguales y solución oral) y se debe monitorear de cerca su estado renal. En pacientes en condiciones que conducen a una reducción del volumen sanguíneo y/o del flujo sanguíneo renal, se debe tener cuidado con las prostaglandinas renales, que desempeńan un papel de apoyo en la mantención de la perfusión renal. En estos pacientes, la administración de AINE puede causar una reducción, que es dosodependiente, en la formación de prostaglandinas renales y puede precipitar lesión renal. Pacientes con alto riesgo de esta reacción son aquellos que presentan disminución de la volemia debido a pérdida de sangre o deshidratación severa, pacientes con insuficiencia renal, con insuficiencia cardíaca, ancianos y aquellos que usan diuréticos. La discontinuación del tratamiento con AINE típicamente continúa con el restablecimiento del estado clínico pretratamiento. El intercambio inadecuado de sangre/líquidos durante la cirugía, que conduce a hipovolemia, puede provocar insuficiencia renal exacerbada cuando se administra ketorolaco trometamol. Por tanto, la pérdida de volumen debe corregirse y la urea y creatinina séricas deben monitorearse rigurosamente. En pacientes en diálisis renal, el aclaramiento de ketorolaco trometamol se redujo a aproximadamente la mitad de la tasa normal y el aumento de la vida media terminal fue de aproximadamente 3 veces. Retención de líquidos y edema: Se han informado con el uso de ketorolaco trometamol y, por tanto, deben usarse con precaución en pacientes con descompensación cardíaca, hipertensión o condiciones similares. Pacientes con insuficiencia hepática: Los pacientes con función hepática alterada debido a cirrosis no deben presentar cambios clínicamente importantes en el aclaramiento de ketorolaco trometamol o en la vida media terminal. Pueden ocurrir elevaciones limítrofes de una o más pruebas de función hepática. Estas anomalías pueden ser transitorias, mantenerse inalteradas o pueden evolucionar con la terapia continua. En estudios clínicos controlados, se produjeron elevaciones significativas (más de tres veces lo normal) en transaminasa piruvato glutamato sérica o en transaminasa oxalacetato glutamato sérica en menos de 1 % de los pacientes. Este medicamento debe ser discontinuado si se presentan signos y síntomas clínicos o manifestaciones sistémicas compatibles con el desarrollo de enfermedad hepática. Efectos hematológicos: Pacientes con trastornos de coagulación sanguínea no deben recibir ketorolaco trometamol. Pacientes en tratamiento anticoagulante pueden presentar un aumento del riesgo de sangrado si se administra simultáneamente ketorolaco trometamol. El uso concomitante de ketorolaco trometamol y una dosis baja profiláctica de heparina (2500 - 5000 unidades cada 12 horas) no se han estudiado ampliamente y también puede estar asociado con aumento del riesgo de sangrado. Pacientes que usan anticoagulantes o que reciben dosis baja de heparina no deben recibir ketorolaco trometamol. Los pacientes que están recibiendo otra terapia con medicamentos que interfieren con la homeostasis deben ser observados cuidadosamente si se administra ketorolaco trometamol. En estudios clínicos controlados, la incidencia clínicamente significativa de sangrado postoperatorio fue menor que 1 %. El ketorolaco trometamol inhibe la agregación plaquetaria y prolonga el tiempo de sangrado. En pacientes con función normal de sangrado, los tiempos se aumentaron, pero no estaban fuera de la tasa normal de 2 a 11 minutos. A diferencia de los efectos prolongados del ácido acetilsalicílico, después de la discontinuación de ketorolaco trometamol, el retorno de la función plaquetaria a la normalidad ocurre dentro de 24 a 48 horas. Se han informado hematomas, epistaxis y otros signos de hemorragia con el uso de este medicamento. Los médicos deben conocer la similitud farmacológica de ketorolaco trometamol con otros fármacos antiinflamatorios no esteroideos que inhiben la ciclooxigenasa y aumentan el riesgo de sangrado, especialmente en ancianos. El riesgo de sangrado gastrointestinal serio es dosodependiente. Esto es particularmente verdadero en pacientes ancianos que recibieron una dosis diaria media máxima de 60 mg/día de ketorolaco trometamol. Este medicamento no es un agente anestésico y no tiene efecto sedante ni propiedades ansiolíticas. Por tanto, el ketorolaco trometamol no debe utilizarse como profilaxis analgésica, como apoyo de anestesia, antes o durante la cirugía o en el postoperatorio en pacientes que presentan alto riesgo de hemorragia u homeostasis incompleta. Se debe tener cuidado cuando la homeostasis es crítica. Efectos sobre sistema nervioso central/sistema musculoesquelético: Sueńos anómalos, pensamientos anómalos, ansiedad, meningitis aséptica, convulsiones, depresión, mareos, somnolencia, sequedad en la boca, euforia, sed excesiva, alucinaciones, cefalea, hipercinesia, incapacidad para concentrarse, insomnio, mialgia, nerviosismo, parestesia, reacciones de tipo psicótico, vértigo. Sistema urinario: Insuficiencia renal aguda, dolor lumbar (con o sin hematuria o uremia), síndrome urémico hemolítico, hiperpotasemia, hiponatremia, aumento de frecuencia urinaria, retención urinaria, nefritis intersticial, síndrome nefrótico, oliguria, aumento de niveles séricos de urea y creatinina. La administración de una dosis de ketorolaco trometamol puede ir seguida de signos indicativos de insuficiencia renal y elevación de los niveles de creatinina y potasio. Órganos de los sentidos: Alteración del gusto, alteración de la visión, zumbidos, pérdida de audición. Piel: Dermatitis exfoliativa, erupción cutánea maculopapular, prurito, urticaria, púrpura, angioedema, sudoración. Reacciones ampollosas que incluyen síndrome de Stevens-Johnson y necrólisis epidérmica tóxica (muy rara). Otros: Astenia, aumento de peso y fiebre. En pacientes mayores de 65 ańos o con menos de 50 kg, no superar la dosis máxima de 40 g/día.
Precauciones:Efectos sobre la capacidad para conducir y utilizar maquinaria: Algunos pacientes pueden presentar mareos, somnolencia, alteraciones visuales, dolor de cabeza, insomnio o depresión con el uso de ketorolaco trometamol. Si los pacientes presentan estos síntomas o efectos secundarios similares, no deben conducir vehículos ni utilizar maquinaria. Pacientes ancianos: Los pacientes mayores de 65 ańos, en comparación con pacientes más jóvenes, pueden presentar un mayor riesgo de eventos adversos. Los riesgos relacionados con edad son comunes para todos los AINE. En comparación con adultos jóvenes, los ancianos tienen una vida media aumentada de ketorolaco trometamol en plasma y un aclaramiento reducido. Embarazo y Lactancia: No hubo evidencia de teratogenicidad en ratas o conejos estudiados con dosis tóxicas maternas de ketorolaco trometamol. En ratas, se ha observado una prolongación del período de gestación y/o un retraso en los partos. Se ha demostrado que ketorolaco trometamol y sus metabolitos pasan al feto y a la leche de los animales. Se ha detectado ketorolaco trometamol en la leche materna en niveles bajos, por lo que no se recomienda la lactancia materna en pacientes que lo estén usando. No se ha establecido la seguridad en el embarazo humano. Se han notificado anomalías congénitas asociadas con la administración de AINE en el hombre, sin embargo, son de baja frecuencia y no siguen ningún patrón discernible. Por tanto, ketorolaco trometamol está contraindicado durante el embarazo, trabajo de parto o en madres lactantes. Categoría de riesgo C: Este medicamento no se debe utilizar por mujeres embarazadas sin orientación médica o del cirujano dentista. Informe inmediatamente a su médico en caso de sospecha de embarazo. Durante el tratamiento, el paciente no debe conducir vehículos u operar máquinas, puesto que su habilidad y atención pueden estar perjudicadas.
Interacciones Medicamentosas:Interacciones medicamento-medicamento: El uso concomitante con otros AINE puede aumentar el riesgo de efectos adversos. Adrenocorticoides, glucocorticoides: Pueden aumentar el riesgo de efectos adversos gastrointestinales. Cumarínicos, indandiónicos, heparina y medicamentos trombolíticos (alteplasa, anistrelasa, estreptoquinasa, uroquinasa): Pueden ser peligrosos debido a la inhibición plaquetaria que ejercen los AINE y también debido al aumento del riesgo de ulceraciones y hemorragias gastrointestinales. Medicamentos inhibidores plaquetarios: Aumentan el riesgo de hemorragia debido al efecto aditivo en la inhibición de la agregación plaquetaria. Cefamandol, cefoperazona, cefotetán, moxalactam o plicamicina: Aumentan el riesgo de úlceras gastrointestinales por sus efectos antiplaquetarios e hipoprotrombinémicos. Antidiabéticos orales o insulina: Aumentan el efecto hipoglicemiante, ya que las prostaglandinas están directamente involucradas en el mecanismo regulador del metabolismo de la glucosa, y también, posiblemente, los antiinflamatorios no esteroideos desplazan a los antidiabéticos orales del complejo proteico plasmático. Antihipertensivos: Hay una reducción o reversión del efecto antihipertensivo, posiblemente debido a la inhibición de las prostaglandinas renales y/o a causar retención de sodio y líquidos. Glucósidos cardíacos: Los AINE pueden exacerbar la insuficiencia cardíaca, reducir la tasa de filtración glomerular y aumentar los niveles plasmáticos de glucósidos cardíacos. Diuréticos: Puede haber una disminución de la eficacia diurética y antihipertensiva y un aumento del riesgo de insuficiencia renal secundaria, probablemente debido a inhibición de la síntesis de prostaglandinas renales. Colchicina: Aumenta el riesgo de hemorragias y ulceraciones gastrointestinales. Compuestos de oro: Comúnmente utilizados en asociación para el tratamiento de artritis pueden aumentar el riesgo de efectos adversos renales. Ciclosporina: Aumenta su concentración sérica por inhibición de las prostaglandinas renales y aumenta el riesgo de nefrotoxicidad. Medicamentos potencialmente depresores medulares o radioterapia: Pueden aumentar el riesgo de efectos adversos hematológicos. Metotrexato: Aumenta la gravedad de los efectos adversos renales. Mifepristona: No se debe administrar ketorolaco trometamol por 8 a 12 días después de su administración, ya que puede reducir sus efectos. Litio: Posiblemente aumenta la concentración sérica de equilibrio del antimaníaco. Probenecid: Aumenta los niveles plasmáticos y la vida media del trometamol. Quinolonas: Aumento del riesgo de presentar convulsiones. Sulfimpirazona: Aumento del riesgo de ulceraciones y hemorragias gastrointestinales. Interacciones Medicamento-Sustancia Química: Evite beber bebidas alcohólicas mientras toma este medicamento.
Sobredosificación: El tratamiento en las primeras horas después de la ingestión consiste en vaciamiento y lavado gástrico o inducción de vómito. El carbón activado podrá administrarse junto con un bloqueador H2. El paciente deberá mantenerse bajo observación y monitoreado en cuanto a la posibilidad de hemorragia gastrointestinal y cambios de las funciones hepática y renal. Se debe implementar un tratamiento de apoyo si es necesario.
Conservación: Este medicamento debe conservarse en su embalaje original, a temperatura ambiente (a no más de 30 °C). Mantener en lugar seco. Número de lote y fechas de fabricación y de caducidad: véase el embalaje. No use medicamento con el plazo de validez caducado. Guárdelo en su embalaje original. Características físicas y organolépticas: Comprimido redondo, biconvexo con marcación en bajo relieve en uno de los lados, de color blanco a amarillo. Antes de usar, observe la apariencia del medicamento. Se deben mantener todos los medicamentos fuera del alcance de los nińos.
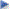 Laboratorio
Laboratorio