Composición: Cada comprimido recubierto contiene: Tegoprazán 50.0 mg. Excipientes: D-manitol, Croscarmelosa Sódica, Hiprolosa, Dióxido de Silicio Coloidal, Estearato de Magnesio, Alcohol Polivinílico, Dióxido de Titanio, Macrogol, Talco, Óxido de Hierro Rojo, Celulosa Microcristalina c.s.
Indicaciones:Ki-Cab está indicado en: Tratamiento de la enfermedad por reflujo gastroesofágico (ERGE) erosiva. Tratamiento de la enfermedad por reflujo gastroesofágico (ERGE) no erosiva. Tratamiento de úlcera gástrica. Terapia antibiótica combinada para la erradicación de Helicobacter pylori en pacientes con úlcera péptica y/o enfermedad gástrica atrófica crónica.
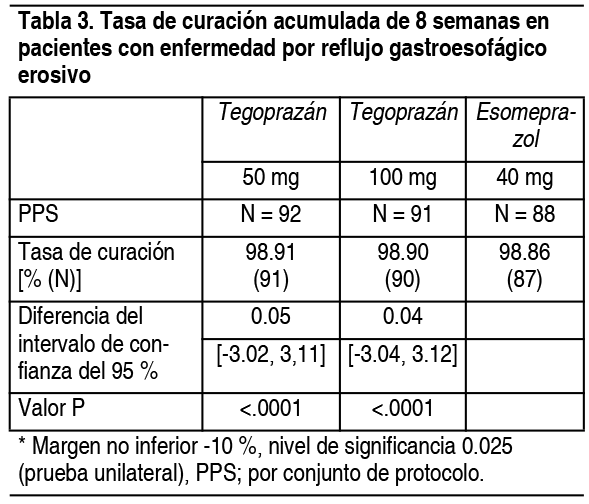
Propiedades:Acción farmacológica: Este fármaco es un bloqueador de ácido competitivo con potasio (P-CAB) que une competitivamente los iones de potasio a la bomba de protones (H+/K+-ATPasa) presente en las células parietales gástricas e inhibe reversiblemente la secreción de ácido en el estómago. Este fármaco se une de manera dependiente de la concentración, inhibe la secreción de ácido gástrico y tiene reversibilidad. Este fármaco inhibe directamente la bomba de protones sin experimentar actividad inducida por ácido. Información farmacocinética: Absorción: Cuando se administró una sola vía oral de 50 a 400 mg de tegoprazán a adultos sanos, el tiempo para alcanzar la concentración máxima en sangre (Tmáx) fue similar en el rango de 0.5 a 1 hora. Después de una sola administración, la concentración plasmática máxima media (Cmáx) y el grado de exposición medio (AUC) de este medicamento tendieron a aumentar en proporción a la dosis dentro del rango de dosis. Después de la administración repetida durante 7 días, la concentración sanguínea máxima promedio de cada grupo de dosis fue similar o disminuyó en comparación con la administración única. Como resultado de la evaluación del efecto dietético sobre la biodisponibilidad después de la administración oral de tegoprazán 200 mg a adultos sanos con el estómago vacío y después de las comidas, se observó un retraso en la Tmáx y una tendencia a disminuir la Cmáx después de comer, pero el AUClast y en los criterios de valoración farmacodinámicos (dentro de estómago el tiempo de retención de pH por encima de 4) no hubo una diferencia significativa. Distribución: La proporción de fármaco que no se une a proteínas in vitro en el tiempo de retención de pH por encima de 4 fue de 8.7 ~ 9.0 % en humanos en el rango de concentración de 1 ~ 10 μm. Metabolismo y excreción: Este fármaco fue metabolizado principalmente por CYP3A4, y se confirmó que el metabolito principal es el metabolito M1 (desalquilado). Tras la administración intravenosa de este fármaco a ratas y perros, la cantidad de excreción corporal inalterada en la orina fue inferior al 1 %. Tras la administración oral de la etiqueta 14C del fármaco en ratas, la tasa de recuperación de excretas a las 168 horas fue del 93 % y 97 % en hembras y machos, respectivamente, 22-24 % y 65-69 % en orina y heces, respectivamente. La intubación biliar fue se excreta en 41.4 %, 25.7 % y 28.4 % en bilis, orina y heces, respectivamente, y la tasa de recuperación total fue del 97.7 % cuando se administró por vía oral a ratas intubadas biliares. Menos del 1 % del tegoprazán sin cambios se encontró en los ácidos biliares y la orina, el 15 % en las heces. El 6 % del metabolito M1 se encontró en las heces. Cuando este fármaco se administró por vía oral a machos adultos sanos, la vida del organismo inalterado y del metabolito M1 fue de 4.1 horas y 22.8 horas, respectivamente. La tasa de excreción de orina es de aproximadamente el 4.1 % y la tasa de pérdida es de aproximadamente 1.1 l/hr. La tasa de excreción urinaria del metabolito principal M1 fue de aproximadamente 2.3 % y la tasa de desaparición fue de 0.5 l/hr. Interacciones farmacológicas: Medicamentos que pueden afectar la concentración plasmática de este medicamento. Este fármaco es metabolizado por CYP3A4 en el hígado. En un estudio in vitro, el ketoconazol, inhibidor de CYP3A4, inhibió significativamente el metabolismo de este fármaco, pero los inhibidores de CYP1A2, CYP2C9, CYP2C19 y CYP2D6 no redujeron significativamente el metabolismo de este fármaco. La coadministración de este medicamento e inhibidores de CYP3A4 puede aumentar la exposición a este medicamento. Este fármaco es un sustrato de P-gp, y el verapamilo, un inhibidor de P-gp, redujo su proporción de reflujo en estudios in vitro. La coadministración de este medicamento con un inhibidor de la P-gp puede aumentar la absorción de este medicamento a través del tracto gastrointestinal, lo que aumenta la exposición. Como resultado de la coadministración de este medicamento y claritromicina (un sustrato e inhibidor de CYP3A4 y P-gp) a adultos sanos, las Css,max y AUC0-12 de este medicamento aumentaron en 1.65 y 2.5 veces, respectivamente. Por otro lado, la claritromicina solo aumentó ligeramente el AUC0-12 en 1.25 veces, no hubo un aumento significativo en Css,max y no se observaron reacciones adversas clínicamente significativas ni reacciones adversas a los medicamentos. Como resultado de la coadministración de este fármaco con metronidazol, tetraciclina y bismuto en adultos sanos, el AUC0-12 de tegoprazán se redujo 0.78 veces y el Css,max 0.75 veces en comparación con la administración de tegoprazán solo y, aunque el AUC0-12 de M1 metabólico tegoprazán disminuyó 0.77 veces y Css,max 0.84 veces, no se observaron reacciones adversas clínicamente significativas ni reacciones adversas al medicamento. Fármacos cuya concentración plasmática puede verse alterada por este fármaco: Este fármaco mostró efectos inhibidores competitivos contra CYP2C8 y CYP3A4 in vitro, pero sus valores de IC50 fueron aproximadamente 25 veces más altos que la concentración plasmática máxima en la dosis clínica. Este fármaco muestra una diferencia en el grado de inhibición según el tipo de sustrato de OATP1B1 in vitro. Considerando la concentración plasmática más alta en la dosis clínica, es posible aumentar ligeramente la concentración en sangre de algunos fármacos sustrato de OATP1B1. Como resultado de la coadministración de este fármaco con metronidazol, tetraciclina y bismuto en adultos sanos, no se afectó nada en la farmacodinamia de metronidazol en comparación con la administración de la coadministración de este fármaco con metronidazol, tetraciclina y bismuto. La AUC0-6 de tetraciclina se redujo 0.62 veces y el Css,max 0.64 veces en comparación, y aunque el AUC0-6 de bismuto aumentó 1.55 veces y Css,max 1.38 veces, no se observaron reacciones adversas clínicamente significativas ni reacciones adversas al medicamento. Información de ensayos clínicos: Enfermedad erosiva por reflujo gastroesofágico: Se realizó un ensayo clínico de fase 3 comparativo, doble ciego, aleatorizado en 302 pacientes con enfermedad por reflujo gastroesofágico erosivo, en el que se administraron 50 mg o 100 mg de este fármaco o 40 mg de esomeprazol durante un máximo de 8 semanas. Como resultado de la prueba, la tasa de curación acumulada a las 8 semanas fue del 98.91 % (91/92 pacientes), 98.90 % (90 pacientes, 91 pacientes), 98.86 % (87 pacientes, 88 pacientes) en el grupo de 50 mg, 100 mg y grupos de tratamiento con esomeprazol 40 mg, respectivamente, que demostró ser no inferior (Tabla 3).
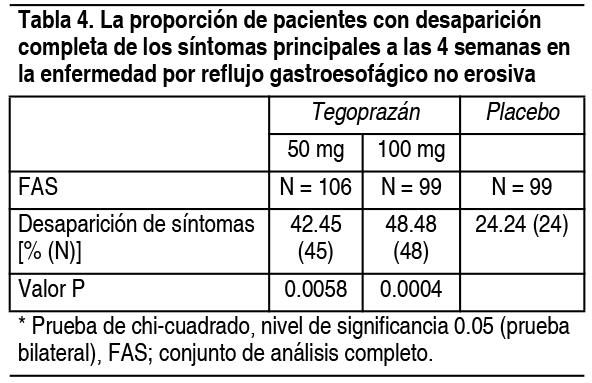
Enfermedad por reflujo gastroesofágico no erosiva: Se realizó un ensayo clínico de fase 3 comparativo, doble ciego, aleatorizado en 324 pacientes con enfermedad por reflujo gastroesofágico no erosivo en el que se administraron 50 mg o 100 mg de este fármaco o placebo durante 4 semanas. Como resultado de la prueba, la proporción de pacientes con desaparición completa de pirosis y reflujo ácido, los principales síntomas durante 7 días consecutivos a las 4 semanas, en los grupos de 50 mg, 100 mg y placebo respectivamente fue del 42.45 % (45/106 pacientes) y del 48.48 % (48/99 pacientes) y 24.24 % (24 personas/99 personas) demostraron superioridad (Tabla 4).
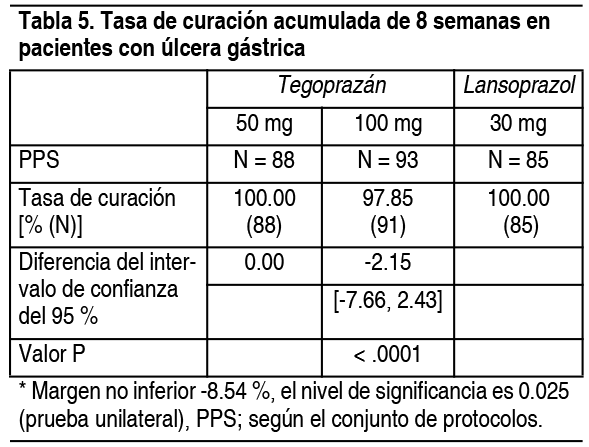
Úlcera gástrica: Se realizó un ensayo clínico de fase 3 comparativo, doble ciego, aleatorizado en 306 pacientes con úlcera gástrica, a los que se les administró 50 mg o 100 mg de este fármaco o 30 mg de lansoprazol durante un máximo de 8 semanas. Como resultado de la prueba, la tasa de curación acumulada a las 8 semanas fue del 100.00 % (88/88), 97.85 % (91/93), 100.00 % (85/85), respectivamente en las dosis de 50 mg, 100 mg y grupos de lansoprazol 30 mg resultó no ser inferior (Tabla 5).
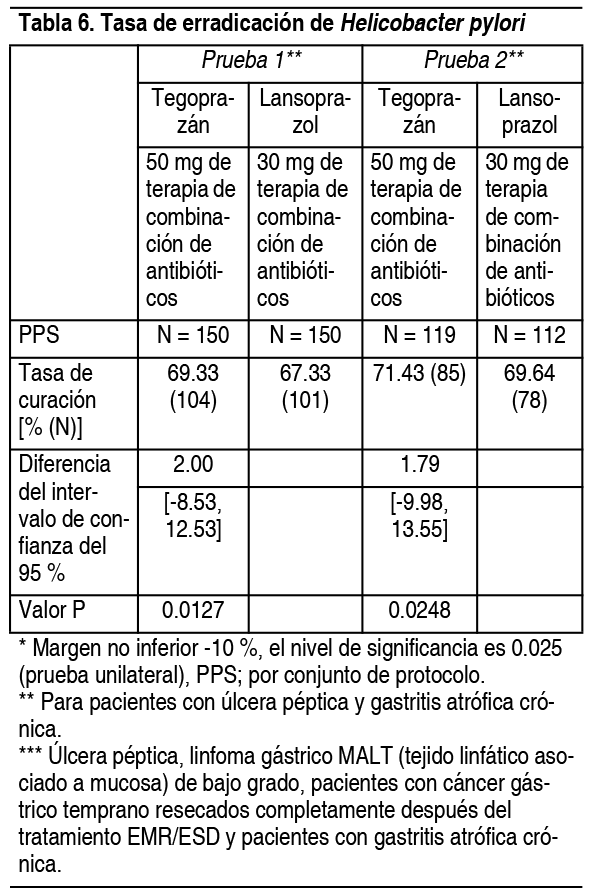
Información sobre pruebas de toxicología: Genotoxicidad: Este fármaco dio negativo en una prueba de mutación inversa con Salmonella y Escherichia coli. La prueba de anomalías cromosómicas in vitro utilizando la línea celular CHL fue positiva, pero la prueba de micronúcleos in vivo utilizando células de médula ósea de rata no indujo micronúcleos, por lo que fue negativa. Carcinogenicidad: Como resultado de una prueba de carcinogenicidad de 2 ańos en ratas, este fármaco se administró en el estómago en machos 15 mg/kg/día (aproximadamente 4.8 veces de la dosis clínica AUC) y 6 mg/kg/día en hembras (aproximadamente 6.8 veces de la dosis clínica AUC) se observaron tumores neuroendocrinos. Toxicidad para la reproducción y el desarrollo: Como resultado de las pruebas de fertilidad y desarrollo embrionario temprano en ratas, no se observó ningún efecto sobre la fertilidad y el desarrollo embrionario temprano hasta una dosis alta de 500 mg/kg/día. Como resultado de la prueba de desarrollo embriofetal, el exceso de costillas cortas del cuello aumentó en 100 ratas y fetos a los que se les administró 500 mg/kg/día. Se determinó que la cantidad de no toxicidad (NOAEL) para la madre de la rata era 500 mg/kg/día, 369 veces la dosis clínica AUC y 20 mg/kg/día para el feto y 15.6 veces la dosis clínica AUC. La dosis máxima (10 mg/kg/día) de conejos presentó síntomas de aborto espontáneo y pérdida de peso, pero no tuvo efecto sobre el desarrollo fetal. Se confirmó que la dosis AUC se duplicó, la cantidad no tóxica (NOAEL) para el embrión y el feto fue de 10 mg/kg/día y 4.8 veces el AUC de la dosis clínica. Como resultado de las pruebas de evaluación de la función materna y prenatal en ratas, se confirmó que el tegoprazán y el metabolito M1 se transfirieron a la leche materna. Se determinó que la cantidad de no toxicidad (NOAEL) era de 20 mg/kg/día y se confirmó que era 8 veces la dosis clínica AUC. Terapia de combinación de antibióticos para la erradicación de H. pylori en pacientes con úlcera péptica y/o gastritis atrófica crónica: En pacientes con úlcera péptica positiva para Helicobacter pylori y gastritis atrófica crónica, se administraron 50 mg de este fármaco o 30 mg de lansoprazol en combinación con el antibiótico amoxicilina 1 g, claritromicina 500 mg 2 veces al día durante 7 días, aleatorizado, doble ciego y comparado, se realizaron 2 ensayos clínicos de fase 3. La tasa de erradicación de Helicobacter pylori no fue inferior en el grupo de terapia combinada con antibióticos de 50 mg de este fármaco y en el grupo de terapia combinada con antibióticos de 30 mg de lansoprazol, respectivamente (Tabla 6).
Posología:Este medicamento se administra a adultos de la siguiente manera: 1. Tratamiento de la enfermedad por reflujo gastroesofágico (ERGE) erosiva: Administrar 1 dosis de 50 mg de Ki-Cab comprimidos recubiertos por vía oral 1 vez al día, durante 4 semanas. Para pacientes con esofagitis no tratada o si los síntomas persisten, administrar durante 4 semanas más. 2. Tratamiento de la enfermedad por reflujo gastroesofágico (ERGE) no erosiva: Administrar 1 dosis de 50 mg de Ki-Cab comprimidos recubiertos por vía oral 1 vez al día, durante 4 semanas. 3. Tratamiento de la úlcera gástrica: Administrar 1 dosis de 50 mg de Ki-Cab comprimidos recubiertos por vía oral 1 vez al día, durante 8 semanas. 4. Combinación de antibióticos para la erradicación de Helicobacter pylori en pacientes con úlcera péptica y/o gastritis atrófica crónica: Los pacientes infectados por Helicobacter pylori deben ser tratados con terapia de desinfección. Se administran 50 mg de este fármaco, 1 g de amoxicilina y 500 mg de claritromicina por vía oral 2 veces al día durante 7 días. Este medicamento se puede administrar con o sin comidas.
Efectos Colaterales: Se realizaron un total de 5 ensayos clínicos en pacientes con enfermedad por reflujo gastroesofágico erosiva, enfermedad por reflujo gastroesofágico no erosiva y úlcera gástrica. De los sujetos que participaron en el ensayo clínico, 360 recibieron 50 mg de este medicamento. Las reacciones adversas y las reacciones adversas a los medicamentos (marcadas con *) notificadas en los ensayos clínicos son las siguientes. Las reacciones adversas (más del 1 %) notificadas comúnmente en este grupo de administración de fármacos se muestran en la Tabla 1 a continuación. Tabla 1: Reacciones adversas notificadas más del 1 % en ensayos clínicos (%). Sistema gastrointestinal: Náusea. Diarrea. Mala digestión. Infección e infecciones parasitarias: Nasofaringitis. Infección por virus del tracto respiratorio superior. Enfermedad sistémica y sitio de administración anormal: Malestar torácico. Otras reacciones adversas informadas en ensayos clínicos con una incidencia de menos del 1 % después de la administración de 50 mg de este medicamento son las siguientes: Trastornos gastrointestinales: Dolor abdominal (superior)*, malestar abdominal*, estreńimiento*, dolor abdominal*, distensión abdominal*, vómitos, eructos, dolor abdominal (inferior), úlcera gástrica*, sangrado rectal*, duodenitis erosiva*, flatulencia*, pólipos gastrointestinales*, enfermedad por reflujo gastroesofágico*, metaplasia epitelial intestinal, hematemesis, hemorroides, heces negras*. Infección e infecciones parasitarias: Foliculitis*, nasofaringitis*, gastroenteritis bacterial, tuberculosis latente. Medidas de laboratorio clínico: Aumento de ALT*, aumento de AST*, aumento de GGT*, aumento de bilirrubina, aumento de CPK*, sangre en orina*, glóbulos rojos positivos en orina*, aumento de gastrina en sangre*, aumento de triglicéridos en sangre*. Enfermedad sistémica y lugar de administración anormal: Fatiga*. Lesiones, intoxicaciones y complicaciones progresivas: Esguince de ligamentos, conmoción cerebral, excoriación, fractura de pie, lesión articular, distensión muscular. Enfermedades musculoesqueléticas y del tejido conjuntivo: Dolores musculares*, articulares, tendinitis*. Trastornos del sistema nervioso: Dolor de cabeza*, mareos. Enfermedades de la piel y del tejido subcutáneo: Angioedema, dermatitis, dermatitis seborreica*. Respiración, pecho, enfermedades mediastínicas: Tos*, dolor de garganta, irritación de garganta. Enfermedades genitales y mamarias: Secreciones vaginales, prurito vulvar, calcificación mamaria*, adenomioma, quiste ovárico. Sistema hepatobiliar: cálculo biliar, quiste hepático. Enfermedades del sistema urinario: Vejiga hiperactiva, nocturia*, quiste renal. Tumores benignos, malignos y no especificados: Adenocarcinoma gástrico, cáncer de mama, adenoma gastrointestinal*, mioma benigno uterino. Enfermedad cardíaca: Latido ventricular prematuro*. Enfermedades de la sangre y del sistema linfático: Linfadenitis*, anemia*. Enfermedad mental: Insomnio*. Implantes dentales de cirugía y tratamiento. Enfermedad del oído y del laberinto: Dolor de oído*. Anomalías metabólicas y nutricionales: Diabetes. Anormalidades cardiovasculares: Hipertensión arterial. Anomalías endocrinas: Quiste tiroideo*. Se realizaron un total de 2 ensayos clínicos en pacientes con úlcera péptica positiva para Helicobacter pylori y gastritis atrófica crónica. De los sujetos que participaron en el ensayo clínico, 314 recibieron 50 mg de este medicamento, 1 g de amoxicilina y 500 mg de claritromicina. Las reacciones adversas y las reacciones adversas a medicamentos notificadas en los ensayos clínicos (*) están marcadas de la siguiente manera. Las reacciones adversas (más del 1 %) notificadas comúnmente en este grupo de administración de medicamentos) se muestran en la Tabla 2 a continuación. Tabla 2: Reacciones adversas notificadas más del 1 % en ensayos clínicos. Sistema gastrointestinal: Diarrea*, dolor abdominal en la parte superior*, distensión abdominal*, indigestión*, náuseas*, dolor abdominal*. Trastornos del sistema nervioso: Trastornos del gusto*, dolor de cabeza*. Enfermedades de la piel y del tejido subcutáneo: Urticaria*. En los ensayos clínicos, otras reacciones adversas notificadas con una incidencia de menos del 1 % después de la administración de 50 mg de este medicamento, amoxicilina 1 g y claritromicina 500 mg en los ensayos clínicos son las siguientes: Enfermedades gastrointestinales: Estreńimiento*, sequedad bucal*, incomodidad abdominal*, incontinencia anal*, duodenitis, heces con sangre, parestesia oral*, vómitos. Trastorno del sistema nervioso: Vértigo*, migrańa*, sueńo*, trastornos del gusto*. Enfermedades de la piel y del tejido subcutáneo: Prurito*, eritema*, erupción*, erupción por medicamentos*, erupción cutánea tóxica*. Infección e infecciones parasitarias: Nasofaringitis, cistitis, herpes zóster, foliculitis*, orzuelo, sinusitis, amigdalitis*. Resultados de laboratorio clínico medidos: Aumento de la enzima activadora del fosfato de creatina en sangre*, aumento de la aminotransferasa del ácido aspártico, aumento de los triglicéridos en sangre, aumento de la lactato deshidrogenasa en sangre*. Trastornos sistémicos y afecciones en el lugar de administración: Astenia*, dolor en el pecho*. Trastornos musculoesqueléticos y del tejido conectivo: Dolor de espalda, mialgia*, rigidez musculoesquelética*. Sistema respiratorio, tórax, enfermedades mediastínicas: Trastorno del habla, tos, dolor orofaríngeo. Enfermedad cardíaca: Palpitaciones*. Enfermedad vascular: Rubor*, enrojecimiento facial*. Diversos trastornos oculares: Coroiditis*, trastornos de la retina*. Diversos trastornos sistémicos: Insomnio*. Neoplasias benignas, malignas y no especificadas (incluidos quistes y pólipos): Adenoma de colon. Trastornos hepatobiliares: Esteatosis hepática. Trastornos metabólicos y nutricionales: Diabetes tipo 2.
Contraindicaciones:No administrar a los siguientes pacientes: Pacientes con reacción de hipersensibilidad y antecedentes de este fármaco, sus componentes o benzimidazoles. Pacientes que toman un medicamento que contenga atazanavir, nelfinavir o rilpivirina. Mujeres embarazadas y lactantes. Administrar con cuidado al siguiente paciente: Pacientes con insuficiencia hepática: No hay experiencia en el uso en pacientes con insuficiencia hepática. En caso de alteración de pruebas de función hepática, se recomienda control y seguimiento. Pacientes con insuficiencia renal (sin experiencia de uso): En caso de alteración de pruebas de función renal, se recomienda control y seguimiento. Ancianos.
Precauciones: Hay síntomas de advertencia (pérdida de peso significativa no intencionada, vómitos recurrentes, disfagia, hematemesis, heces negras, etc.) que sugieren un tumor maligno, ya que este medicamento puede aliviar los síntomas de neoplasias malignas o retrasar el diagnóstico. Si hay una úlcera de estómago o sospechado, debe confirmarse que no es maligno y administrarse. Deficiencia de cianocobalamina (vitamina B12): Si se administra un inhibidor del ácido gástrico a diario durante un período prolongado (p. ej., 3 ańos o más), puede producirse una malabsorción de cianocobalamina debido al ácido hipohidroclórico o la aclorhidria. En la literatura, la deficiencia de cianocobalamina rara vez se ha informado en la administración de inhibidores del ácido gástrico. Este diagnóstico debe considerarse si se observan síntomas clínicos, como deficiencia de cianocobalamina. En caso de tratamiento prolongado con este medicamento, los pacientes deben ser examinados periódicamente. Se observaron pólipos gástricos benignos durante la administración prolongada de este fármaco y otros inhibidores de la secreción de ácido gástrico competitivos con el potasio. Para las úlceras gástricas, etc., la experiencia de uso a largo plazo no es suficiente, por lo que no se utiliza como terapia de mantenimiento. Fractura: Varios estudios observacionales publicados han informado que el tratamiento con inhibidores de la bomba de protones (IBP) puede estar asociado con un mayor riesgo de fracturas de cadera, muńeca y columna relacionadas con la osteoporosis. El riesgo de fractura aumentó en pacientes que recibieron dosis altas de inhibidores de la bomba de protones (dosis con administración diaria repetida) y uso prolongado de más de 1 ańo. Teniendo en cuenta este resultado, este medicamento debe administrarse durante el período de tiempo más corto en una dosis apropiada para la condición de tratamiento. Los pacientes con riesgo de fracturas relacionadas con la osteoporosis deben ser tratados de acuerdo con las pautas de tratamiento establecidas. Rara vez se informó hipomagnesemia en pacientes que habían sido tratados con un inhibidor de la bomba de protones durante más de 3 meses, y los casos más frecuentes se trataron durante más de 1 ańo. En la mayoría de los pacientes, el tratamiento de la hipomagnesemia requiere los suplementos de magnesio y la suspensión del inhibidor de la bomba de protones. Los pacientes que requieran tratamiento a largo plazo o que se administren conjuntamente digoxina o fármacos que causan hipomagnesemia (p. ej., diuréticos) requieren un control periódico de los niveles de magnesio, incluido el inicio del tratamiento. Las reacciones adversas graves incluyen rigidez, arritmia y convulsiones. Cuando la acidez en el estómago disminuye debido a los inhibidores de la bomba de protones, aumenta la cantidad de bacterias presentes generalmente en el tracto gastrointestinal. Cuando se trata con inhibidores del ácido gástrico, el riesgo de infección del tracto gastrointestinal por bacterias como Salmonella, Campylobacter y Clostridium difficile puede aumentar ligeramente. Esto se asocia con un mayor riesgo de diarrea por Clostridium difficile, y varios estudios observacionales han informado que este riesgo aumenta, especialmente en pacientes hospitalizados. Este diagnóstico debe considerarse cuando la diarrea no mejora. Se ha informado diarrea por Clostridium difficile con casi todos los agentes antimicrobianos. Los pacientes deben recibir este medicamento durante el período de tiempo más breve a una dosis adecuada para la afección que se está tratando. No se ha estudiado el efecto de este medicamento sobre la capacidad para conducir o manejar una máquina y no se puede predecir la pérdida de esta capacidad a partir de su acción farmacológica. Sin embargo, al considerar la capacidad de un paciente para operar u operar maquinaria, se deben tener en cuenta la condición clínica del paciente y los aspectos de reacciones adversas de este medicamento. Pólipos de las glándulas fúngicas: El uso de inhibidores de la bomba de protones se asocia con un mayor riesgo de pólipos de las glándulas gástricas, especialmente el uso a largo plazo de más de 1 ańo se asocia con un mayor riesgo de pólipos de las glándulas fúngicas. La mayoría de los pólipos gástricos son asintomáticos. Los inhibidores de la bomba de protones o esta terapia con medicamentos deben usarse durante el período más corto de tiempo a la dosis más baja adecuada al síntoma a tratar. Administración a mujeres embarazadas y lactantes: Mujeres embarazadas: No existen datos de seguridad sobre la exposición humana a este medicamento durante el embarazo. Como resultado de la prueba de desarrollo embriofetal, el exceso de costillas cortas del cuello aumentó en ratas. Por lo tanto, por razones de seguridad, está prohibido el uso de este medicamento durante el embarazo. Madre lactante: No se sabe si este medicamento pasará a la leche materna humana, por lo que no amamante si está tomando este medicamento. Como resultado de las pruebas con animales, se observó que este fármaco se secretaba en la leche cuando se administraba a ratas. Administración en nińos: No se ha establecido la seguridad y eficacia clínicas de este medicamento en nińos y adolescentes. Administración en ancianos: En general, en los ancianos, las funciones fisiológicas como la función hepática o la función renal disminuyen, por lo que debe administrarse con cuidado. Administración a pacientes con insuficiencia renal: No se ha establecido la seguridad y eficacia de este medicamento en pacientes con insuficiencia renal. Administración a pacientes con insuficiencia hepática: No se ha establecido la seguridad y eficacia de este medicamento en pacientes con insuficiencia hepática. Tratamiento en casos de sobredosis: No se han notificado casos de sobredosis grave de este fármaco. En ensayos clínicos, ha habido experiencia de dosis únicas de este fármaco de hasta 400 mg en adultos sanos. En caso de sobredosis, se debe vigilar al paciente para detectar síntomas de toxicidad y, si es necesario, se debe realizar un tratamiento adyuvante general.
Interacciones Medicamentosas:Fármacos que muestran una farmacocinética de absorción dependiente del pH: En el caso de un fármaco cuyo pH ácido gástrico es un determinante importante de la biodisponibilidad, la absorción del fármaco puede inhibirse debido a la supresión de la secreción de ácido gástrico por este fármaco. Al igual que con otros inhibidores de la secreción de ácido y antiácidos, la absorción de ketoconazol, itraconazol, ampicilinester, atazanavir, sales de hierro, erlotinib, gefitinib y micofenolato de mofetilo puede reducirse durante la administración de este medicamento. Por otro lado, se puede incrementar la absorción de fármacos como la digoxina. Dado que este fármaco inhibe la secreción de ácido gástrico, la combinación de atazanavir, nelfinavir o rilpivirina cuya absorción depende del ácido gástrico puede disminuir la concentración plasmática, lo que resulta en una disminución del efecto terapéutico. Por lo tanto, no debe coadministrarse con atazanavir, nelfinavir o rilpivirina. Este fármaco es metabolizado principalmente por CYP3A4. La coadministración de este medicamento y claritromicina, un inhibidor de CYP3A4, aumentó el AUC0-12 de este medicamento y de la claritromicina en 2.5 y 1.25 veces, respectivamente. Este fármaco no tuvo un efecto clínicamente significativo sobre la farmacocinética de la amoxicilina. Este fármaco no afectó la farmacocinética de la atorvastatina. Según un estudio que evaluó la administración concomitante de este fármaco y antiinflamatorios no esteroides (naproxeno, aceclofenaco o celecoxib), no se encontró ninguna interacción farmacocinética clínicamente significativa.
Conservación: Almacenar a no más de 30 °C. Mantener en su envase original, protegido del calor, la luz y la humedad. No usar este producto después de la fecha de vencimiento indicada en el envase. No repita el tratamiento sin antes consultar a su médico. No recomiende este medicamento a otras personas.
 Laboratorio
Laboratorio